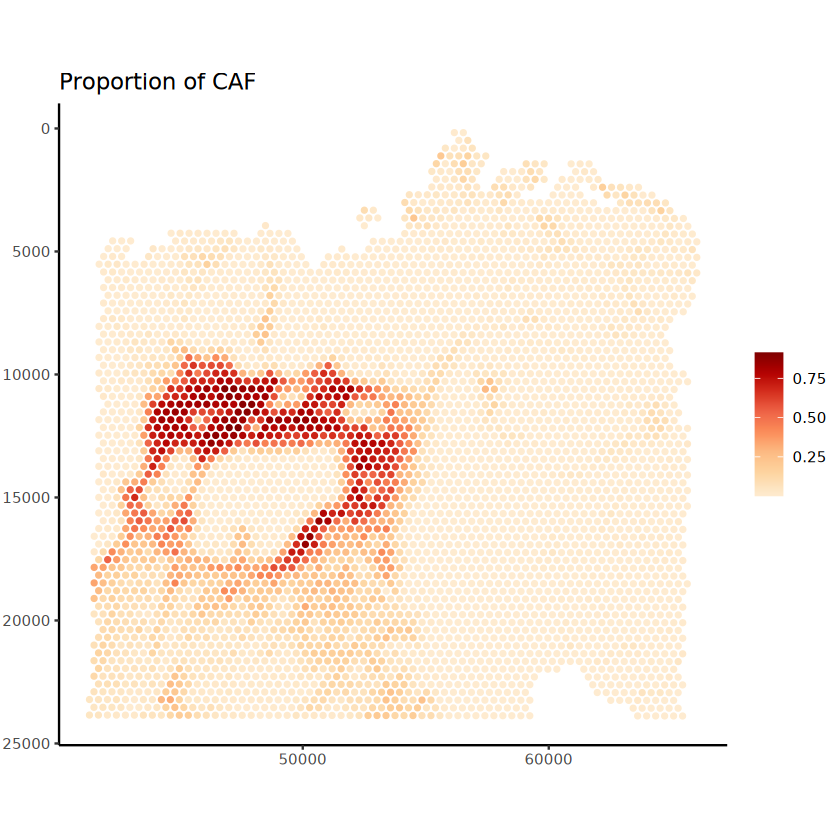
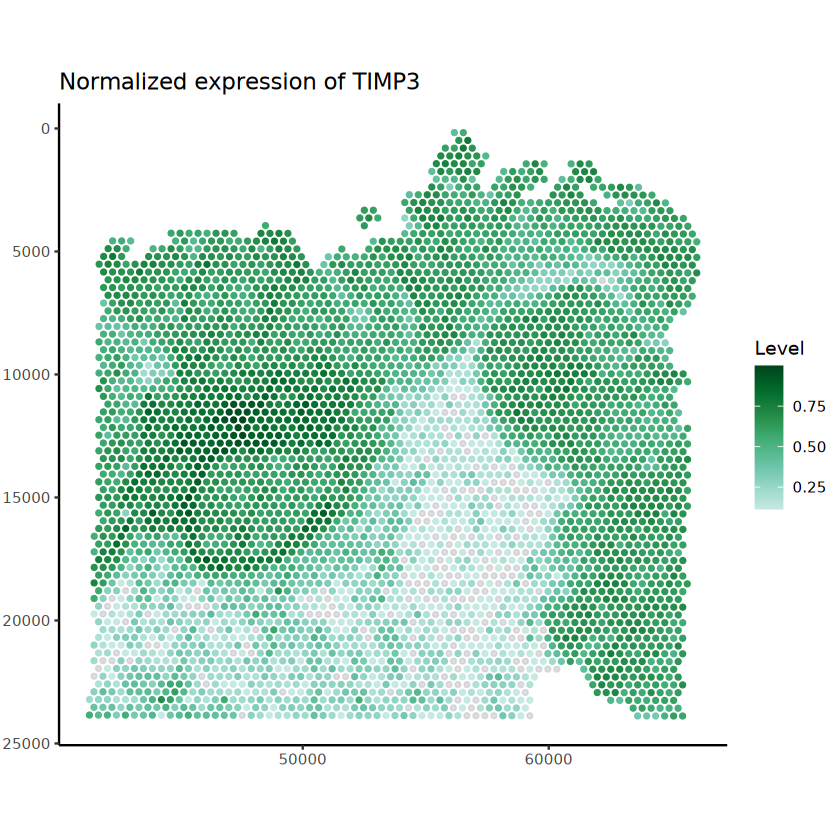
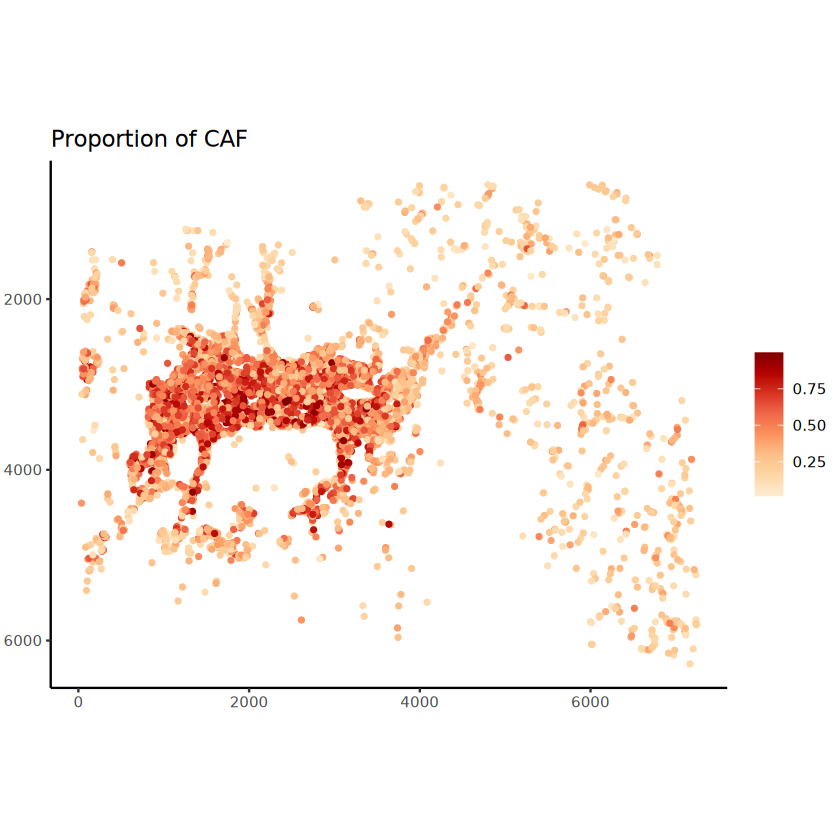
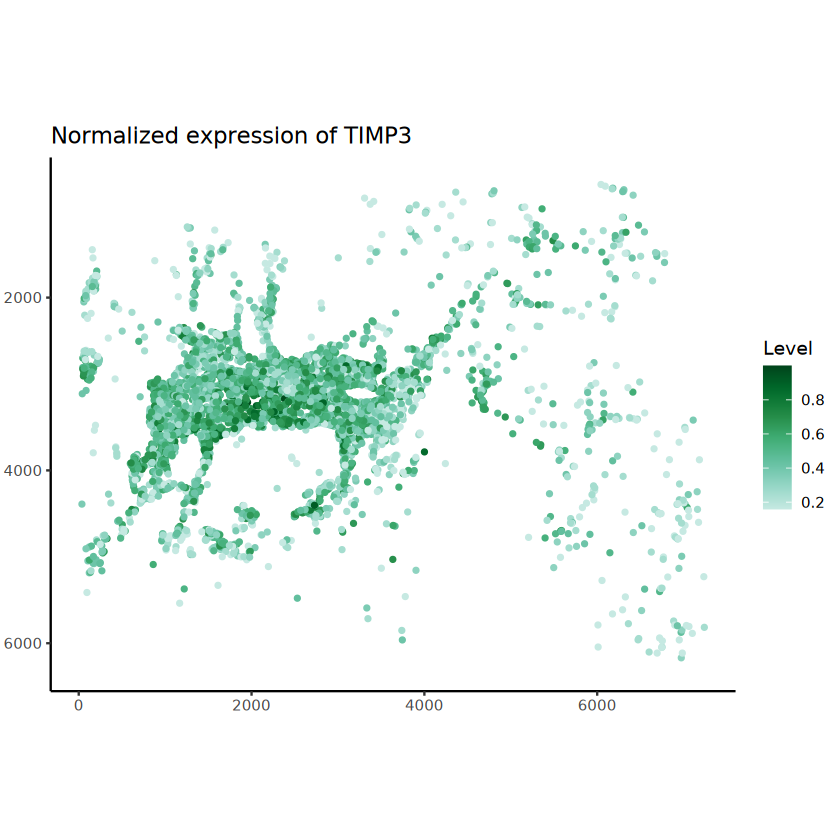
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) CAF, CD4 T cell, Macrophage, Myofibroblast, Pericytes, Plasma, Smooth Muscle, Tumor III, Unknown III (SM) pass the threshold.
Cell type(s) CAF will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 364 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 77 genes to analyze for CAF.
Preprocessed data description: 5000 spots and 32 cell types in total. 5000 spots, 77 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0323293192086494 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.045720561687161 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0323293192086494 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.045720561687161 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for CAF completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) CAF, CD4 T cell, Fibroblast, Macrophage, Myofibroblast, Pericytes, Plasma, Proliferating Immune II, Tumor III, Unknown III (SM) pass the threshold.
Cell type(s) CD4 T cell will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 392 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 87 genes to analyze for CD4 T cell.
Preprocessed data description: 4709 spots and 32 cell types in total. 4709 spots, 87 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0218096971930091 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0308435695616038 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0218096971930091 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0308435695616038 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for CD4 T cell completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) CAF, CD8 Cytotoxic T cell, Macrophage, Tumor III, Unknown III (SM), vSM pass the threshold.
Cell type(s) CD8 Cytotoxic T cell will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 398 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 82 genes to analyze for CD8 Cytotoxic T cell.
Preprocessed data description: 2194 spots and 32 cell types in total. 2194 spots, 82 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0376170009946775 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0531984729824751 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0376170009946775 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0531984729824751 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for CD8 Cytotoxic T cell completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) CAF, Endothelial, Lymphatic Endothelial, Macrophage, Myofibroblast, Pericytes, Plasma, Tumor III pass the threshold.
Cell type(s) Endothelial will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 387 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 104 genes to analyze for Endothelial.
Preprocessed data description: 5000 spots and 32 cell types in total. 5000 spots, 104 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0154691421974095 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0218766706938544 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0154691421974095 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0218766706938544 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Endothelial completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Enteric Glial pass the threshold.
Cell type(s) Enteric Glial will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 391 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 113 genes to analyze for Enteric Glial.
Preprocessed data description: 193 spots and 32 cell types in total. 193 spots, 113 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0221289502402945 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0312950615509038 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0221289502402945 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0312950615509038 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Enteric Glial completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Enterocyte pass the threshold.
Cell type(s) Enterocyte will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 382 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 304 genes to analyze for Enterocyte.
Preprocessed data description: 1318 spots and 32 cell types in total. 1318 spots, 304 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0391229340076264 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0553281838734128 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0391229340076264 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0553281838734128 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Enterocyte completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Fibroblast, Plasma pass the threshold.
Cell type(s) Fibroblast will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 394 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 223 genes to analyze for Fibroblast.
Preprocessed data description: 1173 spots and 32 cell types in total. 1173 spots, 223 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0434392838127099 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0614324243077083 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0434392838127099 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0614324243077083 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Fibroblast completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Goblet, Tumor I, Tumor II, Tumor III, Tumor V pass the threshold.
Cell type(s) Goblet will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 391 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 177 genes to analyze for Goblet.
Preprocessed data description: 5000 spots and 32 cell types in total. 5000 spots, 177 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0277062471579677 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.039182550493259 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0277062471579677 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.039182550493259 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Goblet completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Lymphatic Endothelial pass the threshold.
Cell type(s) Lymphatic Endothelial will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 387 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 170 genes to analyze for Lymphatic Endothelial.
Preprocessed data description: 575 spots and 32 cell types in total. 575 spots, 170 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0252926114667311 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0357691541640844 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0252926114667311 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0357691541640844 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Lymphatic Endothelial completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) CAF, CD4 T cell, Endothelial, Fibroblast, Macrophage, Myofibroblast, Pericytes, Plasma, Proliferating Immune II, Tumor III, Unknown III (SM), vSM pass the threshold.
Cell type(s) Macrophage will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 384 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 75 genes to analyze for Macrophage.
Preprocessed data description: 5000 spots and 32 cell types in total. 5000 spots, 75 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.019734704266815 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0279090864235519 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.019734704266815 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0279090864235519 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Macrophage completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Myofibroblast, Tumor III pass the threshold.
Cell type(s) Myofibroblast will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 389 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 108 genes to analyze for Myofibroblast.
Preprocessed data description: 836 spots and 32 cell types in total. 836 spots, 108 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0424814860461052 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0600778937161653 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0424814860461052 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0600778937161653 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Myofibroblast completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Goblet, Neuroendocrine pass the threshold.
Cell type(s) Neuroendocrine will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 402 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 124 genes to analyze for Neuroendocrine.
Preprocessed data description: 578 spots and 32 cell types in total. 578 spots, 124 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0190330250810503 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0269167622026086 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0190330250810503 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0269167622026086 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Neuroendocrine completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Macrophage, Neutrophil, Tumor III pass the threshold.
Cell type(s) Neutrophil will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 399 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 157 genes to analyze for Neutrophil.
Preprocessed data description: 1713 spots and 32 cell types in total. 1713 spots, 157 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0281073319876069 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0397497700989968 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0281073319876069 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0397497700989968 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Neutrophil completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Endothelial, Macrophage, Myofibroblast, Pericytes, Tumor III pass the threshold.
Cell type(s) Pericytes will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 380 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 95 genes to analyze for Pericytes.
Preprocessed data description: 2832 spots and 32 cell types in total. 2832 spots, 95 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0181779695557007 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0257075310820772 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0181779695557007 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0257075310820772 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Pericytes completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) CAF, Fibroblast, Macrophage, Plasma pass the threshold.
Cell type(s) Plasma will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 397 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 84 genes to analyze for Plasma.
Preprocessed data description: 3261 spots and 32 cell types in total. 3261 spots, 84 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0178435573652251 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0252346008268836 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0178435573652251 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0252346008268836 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Plasma completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) CAF, CD4 T cell, Macrophage, Plasma, Proliferating Immune II pass the threshold.
Cell type(s) Proliferating Immune II will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 390 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 81 genes to analyze for Proliferating Immune II.
Preprocessed data description: 2368 spots and 32 cell types in total. 2368 spots, 81 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.014175889673722 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0200477354352823 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.014175889673722 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0200477354352823 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Proliferating Immune II completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Goblet, Tumor I pass the threshold.
Cell type(s) Tumor I will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 395 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 184 genes to analyze for Tumor I.
Preprocessed data description: 795 spots and 32 cell types in total. 795 spots, 184 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0256831607333036 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0363214742336461 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0256831607333036 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0363214742336461 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Tumor I completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Tumor III pass the threshold.
Cell type(s) Tumor III will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 386 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 359 genes to analyze for Tumor III.
Preprocessed data description: 5000 spots and 32 cell types in total. 5000 spots, 359 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.026347703528003 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0372612796666873 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.026347703528003 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0372612796666873 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Tumor III completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Tumor V pass the threshold.
Cell type(s) Tumor V will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 380 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 126 genes to analyze for Tumor V.
Preprocessed data description: 284 spots and 32 cell types in total. 284 spots, 126 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0189446316493579 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0267917550126845 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0189446316493579 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0267917550126845 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Tumor V completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Unknown III (SM) pass the threshold.
Cell type(s) Unknown III (SM) will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 379 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 99 genes to analyze for Unknown III (SM).
Preprocessed data description: 371 spots and 32 cell types in total. 371 spots, 99 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0683202388756594 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0966194084025271 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0683202388756594 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0966194084025271 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for Unknown III (SM) completed!
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.01 and celltype_threshold = 100.
mcubeFilterCellTypes: Cell type(s) Macrophage, vSM pass the threshold.
Cell type(s) vSM will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 382 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 92 genes to analyze for vSM.
Preprocessed data description: 802 spots and 32 cell types in total. 802 spots, 92 genes, and 1 cell type(s) to analyze.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.0623277404375215 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0881447358388129 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.0623277404375215 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.0881447358388129 for the Gaussian_transformed kernel.
Number of physical cores: 72.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
MCube analysis for vSM completed!