Sentivity and replicability analysis of MCube
We first analyze the senesitivity of MCube to cell type deconvolution using the Visium data. Specifically, we compare the results from MCube when integrting the estimated cell type proportions from two different methods (RCTD and STitch3D). To examine whether the results are replicable, we then compare the results from MCube on the two slices of the Visium data.
[1]:
set.seed(20250428)
library(Matrix)
library(spacexr)
library(MCube)
library(dplyr)
library(ggplot2)
Attaching package: ‘dplyr’
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
[2]:
# RAW_DATA_PATH <- "/import/home/share/zw/data/mouse_brain"
RAW_DATA_PATH <- "/import/home/share/zw/pql/data/mouse_brain"
DATA_PATH <- "/import/home/share/zw/pql/data/mouse_brain/reliability"
RESULT_PATH <- "/import/home/share/zw/pql/results/mouse_brain/reliability"
if (!dir.exists(file.path(DATA_PATH, "visium_1"))) {
dir.create(file.path(DATA_PATH, "visium_1"), recursive = TRUE)
}
if (!dir.exists(file.path(DATA_PATH, "visium_2"))) {
dir.create(file.path(DATA_PATH, "visium_2"), recursive = TRUE)
}
if (!dir.exists(file.path(RESULT_PATH, "visium_1"))) {
dir.create(file.path(RESULT_PATH, "visium_1"), recursive = TRUE)
}
if (!dir.exists(file.path(RESULT_PATH, "visium_2"))) {
dir.create(file.path(RESULT_PATH, "visium_2"), recursive = TRUE)
}
[3]:
# scRNA-seq reference data
sc_counts <- as.data.frame(readr::read_csv(
file.path(RAW_DATA_PATH, "visium_1", "sc_counts.csv")
))
rownames(sc_counts) <- sc_counts[, 1]
sc_counts[, 1] <- NULL
sc_counts <- as.matrix(sc_counts)
dim(sc_counts)
sc_labels <- as.matrix(read.csv(
file.path(RAW_DATA_PATH, "visium_1", "sc_labels.csv"),
header = TRUE, row.names = 1
))[, 1]
# Exclude cells with unknown or low quality labels
# ex_cells <- grep("Unk|LowQ", sc_labels)
ex_cells <- which(sc_labels == "Unk_1")
sc_labels <- sc_labels[-ex_cells]
sc_labels <- factor(sc_labels, levels = unique(sc_labels))
sc_counts <- sc_counts[-ex_cells, ]
reference <- Reference(t(sc_counts), sc_labels, rowSums(sc_counts))
New names:
• `` -> `...1`
Rows: 40532 Columns: 6416
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): ...1
dbl (6415): 0610040J01Rik, 0610043K17Rik, 1110002J07Rik, 1110004F10Rik, 1110...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
- 40532
- 6415
Warning message in Reference(t(sc_counts), sc_labels, rowSums(sc_counts)):
“Reference: number of cells per cell type is 10819, larger than maximum allowable of 10000. Downsampling number of cells to: 10000”
Visium Slice 1
[4]:
counts <- as.data.frame(readr::read_csv(
file.path(RAW_DATA_PATH, "visium_1", "counts.csv")
))
rownames(counts) <- counts[, 1]
counts[, 1] <- NULL
counts <- as.matrix(counts)
dim(counts)
coordinates <- as.data.frame(readr::read_csv(
file.path(RAW_DATA_PATH, "visium_1", "3D_coordinates.csv")
))
rownames(coordinates) <- coordinates[, 1]
coordinates[, c(1, 4)] <- NULL
coordinates <- as.matrix(coordinates)
dim(coordinates)
puck <- SpatialRNA(as.data.frame(coordinates), t(counts), rowSums(counts))
New names:
• `` -> `...1`
Rows: 4168 Columns: 6416
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): ...1
dbl (6415): 0610040J01Rik, 0610043K17Rik, 1110002J07Rik, 1110004F10Rik, 1110...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
- 4168
- 6415
New names:
• `` -> `...1`
Rows: 4168 Columns: 4
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): ...1
dbl (3): x, y, z
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
- 4168
- 2
Integrating with RCTD
Cell type deconvolution
[5]:
myRCTD <- create.RCTD(
puck, reference,
CELL_MIN_INSTANCE = 20, max_cores = 8
)
myRCTD <- run.RCTD(myRCTD, doublet_mode = "full")
saveRDS(myRCTD, file.path(RESULT_PATH, "visium_1", "myRCTD.rds"))
Begin: process_cell_type_info
process_cell_type_info: number of cells in reference: 38849
process_cell_type_info: number of genes in reference: 6415
Ext_L23 Oligo_2 Astro_THAL_med Inh_Sst Inh_6
1244 10000 405 640 467
Ext_L5_3 Inh_Meis2_3 Micro Ext_L5_2 Ext_Pir
127 789 1218 166 954
Astro_AMY Inh_2 Inh_1 Inh_Pvalb Inh_Lamp5
498 839 573 804 331
Ext_L25 Nb_2 Ext_Thal_1 Ext_L56 Ext_Unk_2
922 203 1197 1422 205
Ext_Unk_3 Inh_3 Ext_Amy_2 Ext_Thal_2 Ext_L5_1
416 818 797 671 893
OPC_1 Astro_AMY_CTX Ext_Hpc_DG1 Astro_HYPO Inh_4
1015 279 554 451 1389
Inh_Meis2_1 Ext_Hpc_DG2 Ext_Med Inh_Meis2_2 Ext_Hpc_CA1
363 915 70 602 671
Ext_L6B Astro_CTX Ext_Amy_1 Astro_WM Oligo_1
356 443 519 220 613
Astro_STR Ext_L6 Astro_THAL_lat Nb_1 LowQ_2
81 568 332 225 265
Inh_Vip Astro_HPC Ext_ClauPyr Ext_Hpc_CA2 OPC_2
545 261 272 48 300
Inh_5 Ext_Hpc_CA3 Ext_Unk_1 Inh_Meis2_4 Unk_2
118 232 90 193 85
Astro_THAL_hab LowQ_1 Endo
43 110 22
End: process_cell_type_info
create.RCTD: getting regression differentially expressed genes:
get_de_genes: Ext_L23 found DE genes: 112
get_de_genes: Oligo_2 found DE genes: 411
get_de_genes: Astro_THAL_med found DE genes: 353
get_de_genes: Inh_Sst found DE genes: 123
get_de_genes: Inh_6 found DE genes: 195
get_de_genes: Ext_L5_3 found DE genes: 134
get_de_genes: Inh_Meis2_3 found DE genes: 153
get_de_genes: Micro found DE genes: 647
get_de_genes: Ext_L5_2 found DE genes: 103
get_de_genes: Ext_Pir found DE genes: 69
get_de_genes: Astro_AMY found DE genes: 333
get_de_genes: Inh_2 found DE genes: 98
get_de_genes: Inh_1 found DE genes: 86
get_de_genes: Inh_Pvalb found DE genes: 131
get_de_genes: Inh_Lamp5 found DE genes: 166
get_de_genes: Ext_L25 found DE genes: 147
get_de_genes: Nb_2 found DE genes: 364
get_de_genes: Ext_Thal_1 found DE genes: 213
get_de_genes: Ext_L56 found DE genes: 157
get_de_genes: Ext_Unk_2 found DE genes: 151
get_de_genes: Ext_Unk_3 found DE genes: 151
get_de_genes: Inh_3 found DE genes: 88
get_de_genes: Ext_Amy_2 found DE genes: 74
get_de_genes: Ext_Thal_2 found DE genes: 151
get_de_genes: Ext_L5_1 found DE genes: 106
get_de_genes: OPC_1 found DE genes: 248
get_de_genes: Astro_AMY_CTX found DE genes: 348
get_de_genes: Ext_Hpc_DG1 found DE genes: 196
get_de_genes: Astro_HYPO found DE genes: 352
get_de_genes: Inh_4 found DE genes: 83
get_de_genes: Inh_Meis2_1 found DE genes: 127
get_de_genes: Ext_Hpc_DG2 found DE genes: 184
get_de_genes: Ext_Med found DE genes: 110
get_de_genes: Inh_Meis2_2 found DE genes: 157
get_de_genes: Ext_Hpc_CA1 found DE genes: 186
get_de_genes: Ext_L6B found DE genes: 155
get_de_genes: Astro_CTX found DE genes: 330
get_de_genes: Ext_Amy_1 found DE genes: 113
get_de_genes: Astro_WM found DE genes: 315
get_de_genes: Oligo_1 found DE genes: 358
get_de_genes: Astro_STR found DE genes: 343
get_de_genes: Ext_L6 found DE genes: 94
get_de_genes: Astro_THAL_lat found DE genes: 328
get_de_genes: Nb_1 found DE genes: 239
get_de_genes: LowQ_2 found DE genes: 158
get_de_genes: Inh_Vip found DE genes: 119
get_de_genes: Astro_HPC found DE genes: 337
get_de_genes: Ext_ClauPyr found DE genes: 134
get_de_genes: Ext_Hpc_CA2 found DE genes: 154
get_de_genes: OPC_2 found DE genes: 359
get_de_genes: Inh_5 found DE genes: 172
get_de_genes: Ext_Hpc_CA3 found DE genes: 135
get_de_genes: Ext_Unk_1 found DE genes: 182
get_de_genes: Inh_Meis2_4 found DE genes: 166
get_de_genes: Unk_2 found DE genes: 172
get_de_genes: Astro_THAL_hab found DE genes: 378
get_de_genes: LowQ_1 found DE genes: 416
get_de_genes: Endo found DE genes: 648
get_de_genes: total DE genes: 3435
create.RCTD: getting platform effect normalization differentially expressed genes:
get_de_genes: Ext_L23 found DE genes: 281
get_de_genes: Oligo_2 found DE genes: 588
get_de_genes: Astro_THAL_med found DE genes: 596
get_de_genes: Inh_Sst found DE genes: 235
get_de_genes: Inh_6 found DE genes: 392
get_de_genes: Ext_L5_3 found DE genes: 316
get_de_genes: Inh_Meis2_3 found DE genes: 332
get_de_genes: Micro found DE genes: 970
get_de_genes: Ext_L5_2 found DE genes: 268
get_de_genes: Ext_Pir found DE genes: 214
get_de_genes: Astro_AMY found DE genes: 578
get_de_genes: Inh_2 found DE genes: 275
get_de_genes: Inh_1 found DE genes: 289
get_de_genes: Inh_Pvalb found DE genes: 301
get_de_genes: Inh_Lamp5 found DE genes: 336
get_de_genes: Ext_L25 found DE genes: 327
get_de_genes: Nb_2 found DE genes: 719
get_de_genes: Ext_Thal_1 found DE genes: 403
get_de_genes: Ext_L56 found DE genes: 336
get_de_genes: Ext_Unk_2 found DE genes: 318
get_de_genes: Ext_Unk_3 found DE genes: 316
get_de_genes: Inh_3 found DE genes: 267
get_de_genes: Ext_Amy_2 found DE genes: 226
get_de_genes: Ext_Thal_2 found DE genes: 326
get_de_genes: Ext_L5_1 found DE genes: 276
get_de_genes: OPC_1 found DE genes: 432
get_de_genes: Astro_AMY_CTX found DE genes: 599
get_de_genes: Ext_Hpc_DG1 found DE genes: 376
get_de_genes: Astro_HYPO found DE genes: 591
get_de_genes: Inh_4 found DE genes: 303
get_de_genes: Inh_Meis2_1 found DE genes: 323
get_de_genes: Ext_Hpc_DG2 found DE genes: 361
get_de_genes: Ext_Med found DE genes: 299
get_de_genes: Inh_Meis2_2 found DE genes: 349
get_de_genes: Ext_Hpc_CA1 found DE genes: 324
get_de_genes: Ext_L6B found DE genes: 333
get_de_genes: Astro_CTX found DE genes: 545
get_de_genes: Ext_Amy_1 found DE genes: 296
get_de_genes: Astro_WM found DE genes: 566
get_de_genes: Oligo_1 found DE genes: 524
get_de_genes: Astro_STR found DE genes: 577
get_de_genes: Ext_L6 found DE genes: 286
get_de_genes: Astro_THAL_lat found DE genes: 570
get_de_genes: Nb_1 found DE genes: 523
get_de_genes: LowQ_2 found DE genes: 309
get_de_genes: Inh_Vip found DE genes: 251
get_de_genes: Astro_HPC found DE genes: 572
get_de_genes: Ext_ClauPyr found DE genes: 299
get_de_genes: Ext_Hpc_CA2 found DE genes: 292
get_de_genes: OPC_2 found DE genes: 552
get_de_genes: Inh_5 found DE genes: 391
get_de_genes: Ext_Hpc_CA3 found DE genes: 255
get_de_genes: Ext_Unk_1 found DE genes: 398
get_de_genes: Inh_Meis2_4 found DE genes: 345
get_de_genes: Unk_2 found DE genes: 378
get_de_genes: Astro_THAL_hab found DE genes: 689
get_de_genes: LowQ_1 found DE genes: 812
get_de_genes: Endo found DE genes: 999
get_de_genes: total DE genes: 4497
fitBulk: decomposing bulk
chooseSigma: using initial Q_mat with sigma = 1
Likelihood value: 3620955.45880079
Sigma value: 0.84
Likelihood value: 3547142.55983441
Sigma value: 0.69
Likelihood value: 3487349.56602717
Sigma value: 0.61
Likelihood value: 3461015.8437889
Sigma value: 0.53
Likelihood value: 3439871.52714845
Sigma value: 0.45
Likelihood value: 3425178.73045764
Sigma value: 0.37
Likelihood value: 3418374.87843701
Sigma value: 0.35
Likelihood value: 3418086.29803833
Sigma value: 0.35
[6]:
write.table(
rownames(myRCTD@spatialRNA@counts),
file = file.path(
DATA_PATH, "visium_1",
paste0("HVG_RCTD", ".csv")
),
quote = FALSE, row.names = FALSE, col.names = FALSE
)
[7]:
weights_RCTD <- as.matrix(myRCTD@results$weights)
proportions_RCTD <- weights_RCTD / rowSums(weights_RCTD)
proportions_filter <- proportions_RCTD
proportions_filter[proportions_filter < 0.1] <- 0
major_celltypes <- colnames(proportions_filter)[
which(colSums(proportions_filter) > 50)
]
proportions_RCTD_long_1 <- proportions_RCTD[, major_celltypes] %>%
cbind(myRCTD@spatialRNA@coords) %>%
tidyr::pivot_longer(
cols = -(x:y),
names_to = "Cell type",
values_to = "Proportion"
)
head(proportions_RCTD_long_1)
| x | y | Cell type | Proportion |
|---|---|---|---|
| <dbl> | <dbl> | <chr> | <dbl> |
| 0.274 | 803.368 | Ext_L23 | 1.648713e-05 |
| 0.274 | 803.368 | Oligo_2 | 1.611030e-01 |
| 0.274 | 803.368 | Ext_Pir | 1.648713e-05 |
| 0.274 | 803.368 | Inh_1 | 1.648713e-05 |
| 0.274 | 803.368 | Ext_L25 | 1.648713e-05 |
| 0.274 | 803.368 | Ext_Thal_1 | 1.648713e-05 |
[8]:
p <- ggplot(proportions_RCTD_long_1, aes(x = x, y = y)) +
geom_point(aes(color = Proportion), size = 1, alpha = 1) +
scale_colour_gradientn(name = NULL, colors = pals::brewer.orrd(22)[3:22]) +
scale_y_continuous(trans = "reverse") +
coord_fixed(ratio = 1) +
facet_wrap(~`Cell type`, ncol = 6) +
labs(
title = "Estimated cell type proportions by RCTD",
x = "x", y = "y"
) +
theme_classic() +
theme(
plot.title = element_text(size = 24, hjust = 0.5),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, "visium_1", "proportions_RCTD.pdf"),
plot = p, width = 16, height = 9
)
Identifying cell-type-specific SVGs using MCube
[9]:
myRCTD <- readRDS(file.path(RESULT_PATH, "visium_1", "myRCTD.rds"))
spot_names <- colnames(myRCTD@spatialRNA@counts)
weights_RCTD <- as.matrix(myRCTD@results$weights)
spot_effects_RCTD <- log(rowSums(weights_RCTD))
names(spot_effects_RCTD) <- rownames(weights_RCTD)
[10]:
mcube_object_RCTD_1 <- createMCube(
counts = t(as.matrix(myRCTD@originalSpatialRNA@counts[, spot_names])),
coordinates = as.matrix(myRCTD@spatialRNA@coords),
proportions = weights_RCTD / rowSums(weights_RCTD),
library_sizes = myRCTD@spatialRNA@nUMI,
reference = t(myRCTD@cell_type_info$info[[1]]),
used_for_deconvolution = rownames(myRCTD@spatialRNA@counts),
spot_effects = spot_effects_RCTD,
celltype_test = grep("Unk|LowQ", levels(sc_labels), value = TRUE, invert = TRUE),
celltype_threshold = 50,
project = "mouse_brain_visium_1"
)
mcube_object_RCTD_1 <- mcubeFitNull(
mcube_object_RCTD_1,
num_workers = 36, num_threads = 1
)
mcube_object_RCTD_1 <- mcubeTest(
mcube_object_RCTD_1,
num_workers = 36, num_threads = 1, shared_memory = TRUE
)
saveRDS(
mcube_object_RCTD_1,
file = file.path(
RESULT_PATH, "visium_1",
paste0("mcube_RCTD", ".rds")
)
)
saveRDS(
mcube_object_RCTD_1@pvalues,
file = file.path(
RESULT_PATH, "visium_1",
paste0("mcube_pvalues_RCTD", ".rds")
)
)
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.1 and celltype_threshold = 50.
mcubeFilterCellTypes: Cell type(s) Ext_L23, Oligo_2, Ext_Pir, Inh_1, Ext_L25, Ext_Thal_1, Ext_L56, Ext_Thal_2, Ext_L5_1, Astro_AMY_CTX, Ext_Hpc_DG1, Astro_HYPO, Inh_4, Ext_Med, Ext_Hpc_CA1, Oligo_1, Astro_HPC pass the threshold.
Cell type(s) Astro_THAL_med, Inh_Sst, Inh_6, Ext_L5_3, Inh_Meis2_3, Micro, Ext_L5_2, Astro_AMY, Inh_2, Inh_Pvalb, Inh_Lamp5, Nb_2, Inh_3, Ext_Amy_2, OPC_1, Inh_Meis2_1, Ext_Hpc_DG2, Inh_Meis2_2, Ext_L6B, Astro_CTX, Ext_Amy_1, Astro_WM, Astro_STR, Ext_L6, Astro_THAL_lat, Nb_1, Inh_Vip, Ext_ClauPyr, Ext_Hpc_CA2, OPC_2, Inh_5, Ext_Hpc_CA3, Inh_Meis2_4, Astro_THAL_hab, Endo has/have less than the minimum celltype_threshold = 50 with proportion_threshold = 0.1.
To include the above cell type(s), please reduce celltype_threshold or proportion_threshold.
Cell type(s) Ext_L23, Oligo_2, Ext_Pir, Inh_1, Ext_L25, Ext_Thal_1, Ext_L56, Ext_Thal_2, Ext_L5_1, Astro_AMY_CTX, Ext_Hpc_DG1, Astro_HYPO, Inh_4, Ext_Med, Ext_Hpc_CA1, Oligo_1, Astro_HPC will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 2768 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 934 genes to analyze for Ext_L23.
mcubeFilterGenesCellType: Select 1056 genes to analyze for Oligo_2.
mcubeFilterGenesCellType: Select 999 genes to analyze for Ext_Pir.
mcubeFilterGenesCellType: Select 1140 genes to analyze for Inh_1.
mcubeFilterGenesCellType: Select 969 genes to analyze for Ext_L25.
mcubeFilterGenesCellType: Select 928 genes to analyze for Ext_Thal_1.
mcubeFilterGenesCellType: Select 1006 genes to analyze for Ext_L56.
mcubeFilterGenesCellType: Select 1017 genes to analyze for Ext_Thal_2.
mcubeFilterGenesCellType: Select 948 genes to analyze for Ext_L5_1.
mcubeFilterGenesCellType: Select 1097 genes to analyze for Astro_AMY_CTX.
mcubeFilterGenesCellType: Select 973 genes to analyze for Ext_Hpc_DG1.
mcubeFilterGenesCellType: Select 1118 genes to analyze for Astro_HYPO.
mcubeFilterGenesCellType: Select 1143 genes to analyze for Inh_4.
mcubeFilterGenesCellType: Select 1018 genes to analyze for Ext_Med.
mcubeFilterGenesCellType: Select 866 genes to analyze for Ext_Hpc_CA1.
mcubeFilterGenesCellType: Select 851 genes to analyze for Oligo_1.
mcubeFilterGenesCellType: Select 1093 genes to analyze for Astro_HPC.
Preprocessed data description: 4167 spots and 58 cell types in total. 3989 spots, 2759 genes, and 17 cell type(s) to analyze.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.101063440730398 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.142925288541018 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.101063440730398 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.142925288541018 for the Gaussian_transformed kernel.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
Integrating with STitch3D
Load in deconvolution results
[11]:
reference_ST <- data.matrix(read.csv(
file.path(DATA_PATH, "visium_1", "reference_ST.csv"),
header = TRUE, row.names = 1, check.names = FALSE
))
dim(reference_ST)
num_celltypes <- nrow(reference_ST)
proportions_ST <- data.matrix(read.csv(
file.path(DATA_PATH, "visium_1", paste0("prop_slice", 0, ".csv")),
header = TRUE, row.names = 1, check.names = FALSE
))
dim(proportions_ST)
counts <- as.data.frame(readr::read_csv(
file.path(DATA_PATH, "visium_1", "counts.csv")
))
rownames(counts) <- counts[, 1]
counts[, 1] <- NULL
counts <- data.matrix(counts)
dim(counts)
coordinates <- data.matrix(read.csv(
file.path(DATA_PATH, "visium_1", "3D_coordinates.csv"),
header = TRUE, row.names = 1, check.names = FALSE
))[, c("x", "y")]
dim(coordinates)
spot_effects_ST <- data.matrix(read.csv(
file.path(DATA_PATH, "visium_1", "spot_effects_ST.csv"),
header = TRUE, row.names = 1, check.names = FALSE
))[, 1]
library_sizes_ST <- data.matrix(read.csv(
file.path(DATA_PATH, "visium_1", "library_sizes_ST.csv"),
header = TRUE, row.names = 1, check.names = FALSE
))[, 1]
- 58
- 4497
- 4168
- 58
New names:
• `` -> `...1`
Rows: 4168 Columns: 4498
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): ...1
dbl (4497): Xkr4, Sox17, Rgs20, Oprk1, Rb1cc1, St18, Sntg1, Adhfe1, Sgk3, Mc...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
- 4168
- 4497
- 4168
- 2
[12]:
proportions_filter <- proportions_ST
proportions_filter[proportions_filter < 0.1] <- 0
major_celltypes <- colnames(proportions_filter)[
which(colSums(proportions_filter) > 50)
]
proportions_ST_long_1 <- as.data.frame(proportions_ST[, major_celltypes]) %>%
cbind(coordinates) %>%
tidyr::pivot_longer(
cols = -(x:y),
names_to = "Cell type",
values_to = "Proportion"
)
head(proportions_ST_long_1)
| x | y | Cell type | Proportion |
|---|---|---|---|
| <dbl> | <dbl> | <chr> | <dbl> |
| 0.274 | 803.368 | Astro_HYPO | 1.185785e-01 |
| 0.274 | 803.368 | Ext_Amy_2 | 9.673296e-07 |
| 0.274 | 803.368 | Ext_Hpc_CA1 | 6.732587e-07 |
| 0.274 | 803.368 | Ext_Hpc_DG1 | 3.355544e-09 |
| 0.274 | 803.368 | Ext_L23 | 8.121780e-10 |
| 0.274 | 803.368 | Ext_L25 | 4.116661e-08 |
[13]:
p <- ggplot(proportions_ST_long_1, aes(x = x, y = y)) +
geom_point(aes(color = Proportion), size = 1, alpha = 1) +
scale_colour_gradientn(name = NULL, colors = pals::brewer.orrd(22)[3:22]) +
scale_y_continuous(trans = "reverse") +
coord_fixed(ratio = 1) +
facet_wrap(~`Cell type`, nrow = 3) +
labs(
title = "Estimated cell type proportions by STitch3D",
x = "x", y = "y"
) +
theme_classic() +
theme(
plot.title = element_text(size = 24, hjust = 0.5),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, "visium_1", "proportions_STitch3D.pdf"),
plot = p, width = 16, height = 9
)
Identifying cell-type-specific SVGs using MCube
[14]:
mcube_object_ST_1 <- createMCube(
counts = counts,
coordinates = coordinates,
proportions = proportions_ST,
library_sizes = library_sizes_ST,
reference = reference_ST,
spot_effects = spot_effects_ST,
celltype_test = grep("Unk|LowQ", levels(sc_labels), value = TRUE, invert = TRUE),
celltype_threshold = 50,
project = "mouse_brain_visium_1"
)
mcube_object_ST_1 <- mcubeFitNull(
mcube_object_ST_1,
num_workers = 36, num_threads = 1
)
mcube_object_ST_1 <- mcubeTest(
mcube_object_ST_1,
num_workers = 36, num_threads = 1, shared_memory = TRUE
)
saveRDS(
mcube_object_ST_1,
file = file.path(
RESULT_PATH, "visium_1",
paste0("mcube_ST", ".rds")
)
)
saveRDS(
mcube_object_ST_1@pvalues,
file = file.path(
RESULT_PATH, "visium_1",
paste0("mcube_pvalues_ST", ".rds")
)
)
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.1 and celltype_threshold = 50.
mcubeFilterCellTypes: Cell type(s) Astro_HYPO, Ext_Amy_2, Ext_Hpc_CA1, Ext_Hpc_DG1, Ext_L23, Ext_L25, Ext_L56, Ext_L5_1, Ext_L5_2, Ext_Pir, Ext_Thal_1, Ext_Thal_2, Ext_Unk_3, Inh_1, Inh_2, Inh_3, Inh_4, Oligo_1, Oligo_2 pass the threshold.
Cell type(s) Astro_AMY, Astro_AMY_CTX, Astro_CTX, Astro_HPC, Astro_STR, Astro_THAL_hab, Astro_THAL_lat, Astro_THAL_med, Astro_WM, Endo, Ext_Amy_1, Ext_ClauPyr, Ext_Hpc_CA2, Ext_Hpc_CA3, Ext_Hpc_DG2, Ext_L5_3, Ext_L6, Ext_L6B, Ext_Med, Inh_5, Inh_6, Inh_Lamp5, Inh_Meis2_1, Inh_Meis2_2, Inh_Meis2_3, Inh_Meis2_4, Inh_Pvalb, Inh_Sst, Inh_Vip, Micro, Nb_1, Nb_2, OPC_1, OPC_2 has/have less than the minimum celltype_threshold = 50 with proportion_threshold = 0.1.
To include the above cell type(s), please reduce celltype_threshold or proportion_threshold.
Cell type(s) Astro_HYPO, Ext_Amy_2, Ext_Hpc_CA1, Ext_Hpc_DG1, Ext_L23, Ext_L25, Ext_L56, Ext_L5_1, Ext_L5_2, Ext_Pir, Ext_Thal_1, Ext_Thal_2, Inh_1, Inh_2, Inh_3, Inh_4, Oligo_1, Oligo_2 will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 2599 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 1083 genes to analyze for Astro_HYPO.
mcubeFilterGenesCellType: Select 921 genes to analyze for Ext_Amy_2.
mcubeFilterGenesCellType: Select 802 genes to analyze for Ext_Hpc_CA1.
mcubeFilterGenesCellType: Select 911 genes to analyze for Ext_Hpc_DG1.
mcubeFilterGenesCellType: Select 875 genes to analyze for Ext_L23.
mcubeFilterGenesCellType: Select 894 genes to analyze for Ext_L25.
mcubeFilterGenesCellType: Select 949 genes to analyze for Ext_L56.
mcubeFilterGenesCellType: Select 896 genes to analyze for Ext_L5_1.
mcubeFilterGenesCellType: Select 853 genes to analyze for Ext_L5_2.
mcubeFilterGenesCellType: Select 926 genes to analyze for Ext_Pir.
mcubeFilterGenesCellType: Select 881 genes to analyze for Ext_Thal_1.
mcubeFilterGenesCellType: Select 972 genes to analyze for Ext_Thal_2.
mcubeFilterGenesCellType: Select 1087 genes to analyze for Inh_1.
mcubeFilterGenesCellType: Select 1006 genes to analyze for Inh_2.
mcubeFilterGenesCellType: Select 1073 genes to analyze for Inh_3.
mcubeFilterGenesCellType: Select 1081 genes to analyze for Inh_4.
mcubeFilterGenesCellType: Select 804 genes to analyze for Oligo_1.
mcubeFilterGenesCellType: Select 991 genes to analyze for Oligo_2.
Preprocessed data description: 4168 spots and 58 cell types in total. 3928 spots, 2587 genes, and 18 cell type(s) to analyze.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.101059327818091 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.142919472004653 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.101059327818091 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.142919472004653 for the Gaussian_transformed kernel.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
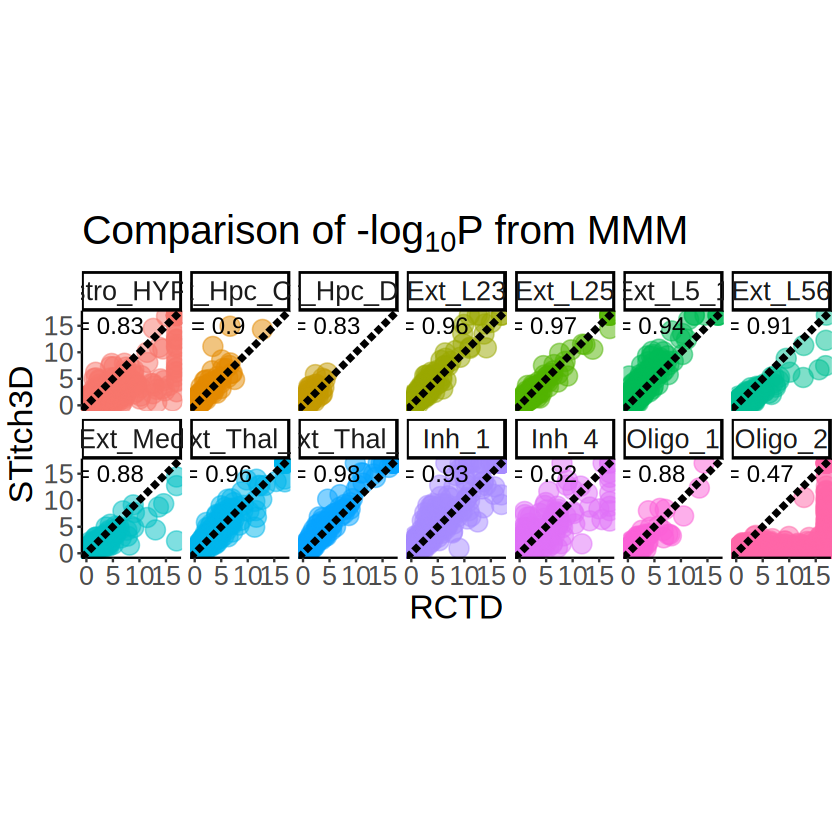
Comparison of the deconvolution results
[15]:
overlap_celltypes <- intersect(
unique(proportions_RCTD_long_1$`Cell type`),
unique(proportions_ST_long_1$`Cell type`)
)
overlap_celltypes <- sort(overlap_celltypes)
overlap_celltypes
- 'Astro_HYPO'
- 'Ext_Hpc_CA1'
- 'Ext_Hpc_DG1'
- 'Ext_L23'
- 'Ext_L25'
- 'Ext_L5_1'
- 'Ext_L56'
- 'Ext_Pir'
- 'Ext_Thal_1'
- 'Ext_Thal_2'
- 'Inh_1'
- 'Inh_4'
- 'Oligo_1'
- 'Oligo_2'
[16]:
p <- ggplot(
proportions_RCTD_long_1[proportions_RCTD_long_1$`Cell type` %in% overlap_celltypes, ],
aes(x = x, y = y)
) +
geom_point(aes(color = Proportion), size = 1, alpha = 1) +
scale_colour_gradientn(name = NULL, colors = pals::brewer.orrd(22)[3:22]) +
scale_y_continuous(trans = "reverse") +
coord_fixed(ratio = 1) +
facet_wrap(~`Cell type`, nrow = 2) +
labs(
title = "Estimated cell type proportions by RCTD",
x = "x", y = "y"
) +
theme_classic() +
theme(
plot.title = element_text(size = 24, hjust = 0.5),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, "visium_1", "sensitivity_proportions_RCTD.pdf"),
plot = p, width = 16, height = 6
)
[17]:
p <- ggplot(
proportions_ST_long_1[proportions_ST_long_1$`Cell type` %in% overlap_celltypes, ],
aes(x = x, y = y)
) +
geom_point(aes(color = Proportion), size = 1, alpha = 1) +
scale_colour_gradientn(name = NULL, colors = pals::brewer.orrd(22)[3:22]) +
scale_y_continuous(trans = "reverse") +
coord_fixed(ratio = 1) +
facet_wrap(~`Cell type`, nrow = 2) +
labs(
title = "Estimated cell type proportions by STitch3D",
x = "x", y = "y"
) +
theme_classic() +
theme(
plot.title = element_text(size = 24, hjust = 0.5),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, "visium_1", "sensitivity_proportions_STitch3D.pdf"),
plot = p, width = 16, height = 6
)
Comparison of the cell-type-specific SVG results
[18]:
mcube_pvalues_RCTD_1 <- readRDS(
file = file.path(
RESULT_PATH, "visium_1",
paste0("mcube_pvalues_RCTD", ".rds")
)
)
mcube_pvalues_ST_1 <- readRDS(
file = file.path(
RESULT_PATH, "visium_1",
paste0("mcube_pvalues_ST", ".rds")
)
)
[19]:
mcube_pvalues_RCTD_1 <- mcubePvalueList2LongTable(mcube_pvalues_RCTD_1)
mcube_pvalues_RCTD_1$minus_log10_pvalue <- -log10(mcube_pvalues_RCTD_1$pvalue)
mcube_pvalues_RCTD_1$minus_log10_pvalue[mcube_pvalues_RCTD_1$minus_log10_pvalue > 17] <- 17
mcube_pvalues_ST_1 <- mcubePvalueList2LongTable(mcube_pvalues_ST_1)
mcube_pvalues_ST_1$minus_log10_pvalue <- -log10(mcube_pvalues_ST_1$pvalue)
mcube_pvalues_ST_1$minus_log10_pvalue[mcube_pvalues_ST_1$minus_log10_pvalue > 17] <- 17
pvalues_merge <- merge(
mcube_pvalues_RCTD_1, mcube_pvalues_ST_1,
by = c("celltype", "gene"),
suffixes = c("_RCTD", "_ST")
)
head(pvalues_merge)
| celltype | gene | pvalue_RCTD | minus_log10_pvalue_RCTD | pvalue_ST | minus_log10_pvalue_ST | |
|---|---|---|---|---|---|---|
| <chr> | <chr> | <dbl> | <dbl> | <dbl> | <dbl> | |
| 1 | Astro_HYPO | 1110004F10Rik | 0.011954503 | 1.9224685 | 0.07614846 | 1.1183389 |
| 2 | Astro_HYPO | 1110051M20Rik | 0.008504576 | 2.0703473 | 0.16657483 | 0.7783906 |
| 3 | Astro_HYPO | 2310022B05Rik | 0.425145644 | 0.3714623 | 0.48544593 | 0.3138591 |
| 4 | Astro_HYPO | 2700081O15Rik | 0.120882778 | 0.9176356 | 0.37604763 | 0.4247571 |
| 5 | Astro_HYPO | 4930402H24Rik | 0.654651260 | 0.1839900 | 0.27306509 | 0.5637338 |
| 6 | Astro_HYPO | 4933431E20Rik | 0.181237154 | 0.7417528 | 0.22063771 | 0.6563203 |
[20]:
correlations <- pvalues_merge %>%
group_by(celltype) %>%
summarize(
correlation = cor(minus_log10_pvalue_RCTD, minus_log10_pvalue_ST, method = "pearson")
)
[21]:
p <- ggplot(pvalues_merge, aes(x = minus_log10_pvalue_RCTD, y = minus_log10_pvalue_ST)) +
geom_point(aes(color = celltype), size = 5, alpha = 0.5) +
geom_abline(slope = 1, intercept = 0, linetype = "longdash", color = "black", linewidth = 1.5) +
geom_text(
data = correlations,
mapping = aes(
x = 4, y = 15,
label = paste("r =", round(correlation, 2))
),
size = 5
) +
scale_x_continuous(limits = c(0, 17), oob = scales::oob_squish) +
scale_y_continuous(limits = c(0, 17), oob = scales::oob_squish) +
coord_fixed(ratio = 1) +
labs(
title = expression(paste("Comparison of ", "-log"[10], plain("P"), " from MMM")),
x = "RCTD", y = "STitch3D"
) +
facet_wrap(celltype ~ ., nrow = 2) +
theme_classic() +
theme(
plot.title = element_text(size = 24),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_text(size = 20),
axis.title.y = element_text(size = 20),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "none"
)
ggsave(
file.path(RESULT_PATH, "visium_1", "MMM_RCTD_vs_STitch3D.pdf"),
plot = p,
width = 15, height = 6
)
ggsave(
file.path(RESULT_PATH, "visium_1", "MMM_RCTD_vs_STitch3D.png"),
plot = p,
width = 15, height = 6
)
p
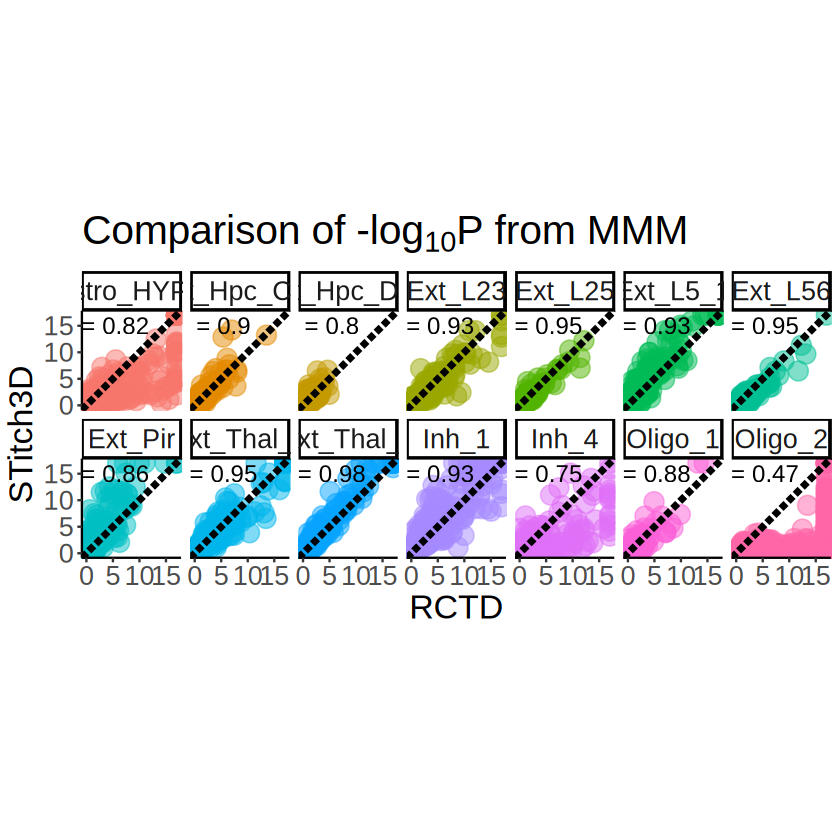
Detailed comparison of Oligo_2
[22]:
# mcube_object_RCTD_1 <- readRDS(
# file = file.path(
# RESULT_PATH, "visium_1",
# paste0("mcube_RCTD", ".rds")
# )
# )
# mcube_object_ST_1 <- readRDS(
# file = file.path(
# RESULT_PATH, "visium_1",
# paste0("mcube_ST", ".rds")
# )
# )
[23]:
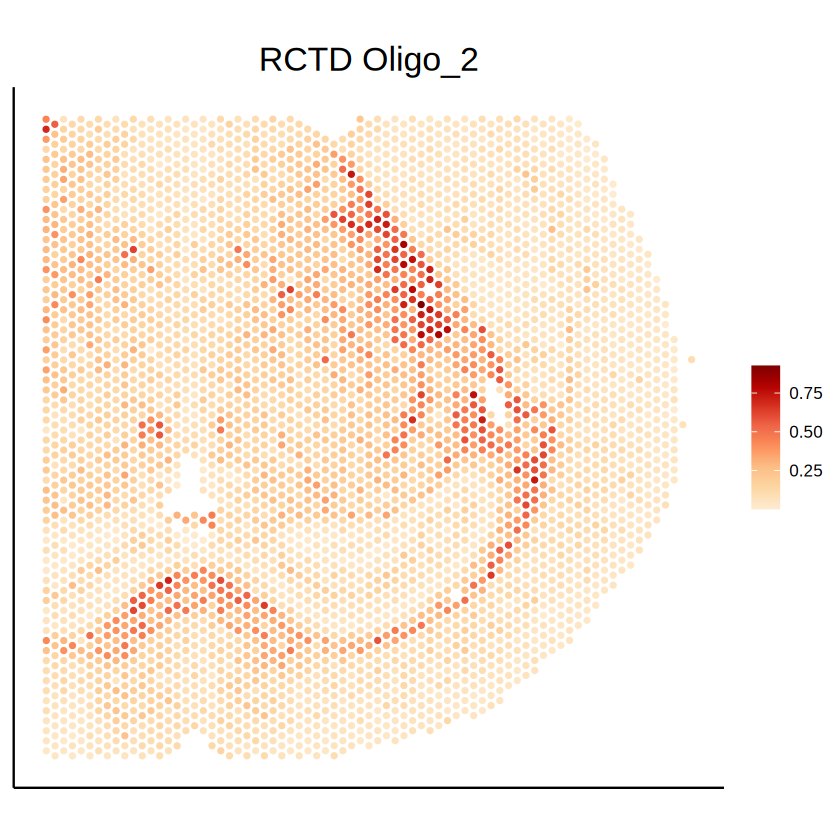
celltype <- "Oligo_2"
p1 <- mcubePlotPropCellType(mcube_object_RCTD_1@proportions, mcube_object_RCTD_1@coordinates, celltype) +
labs(title = paste("RCTD", celltype)) +
theme(
text = element_text(size = 1, hjust = 0.5),
plot.title = element_text(size = 20, hjust = 0.5),
axis.title = element_blank(),
axis.text.x = element_blank(),
axis.text.y = element_blank(),
axis.ticks = element_blank(),
legend.text = element_text(size = 10),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, paste0("proportion_", celltype, "_RCTD", ".pdf")),
plot = p1, width = 4, height = 4
)
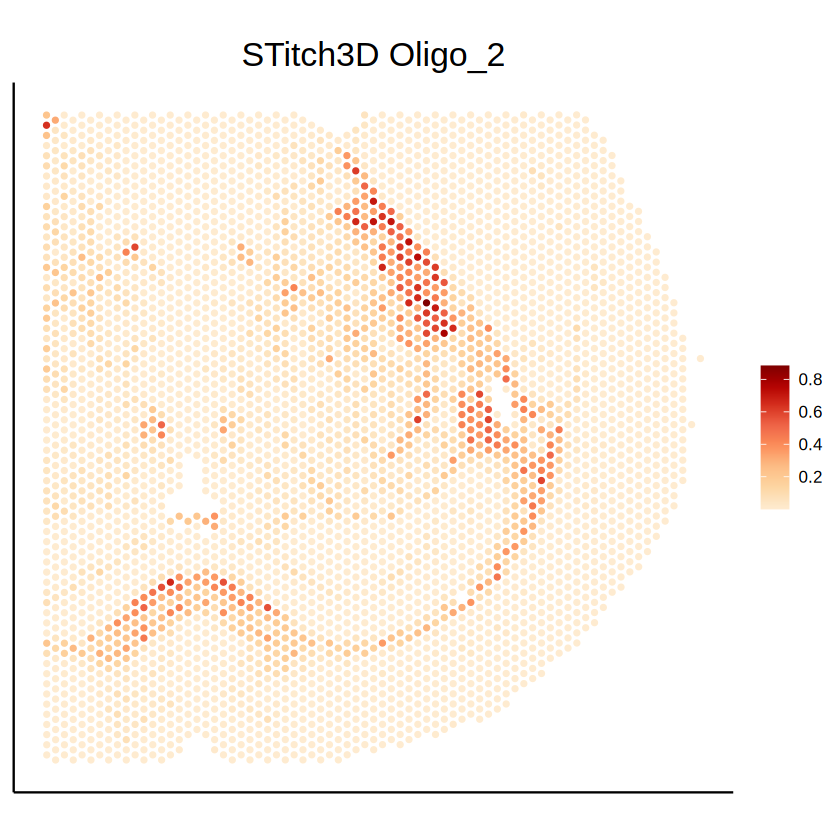
p2 <- mcubePlotPropCellType(mcube_object_ST_1@proportions, mcube_object_ST_1@coordinates, celltype) +
labs(title = paste("STitch3D", celltype)) +
theme(
text = element_text(size = 1, hjust = 0.5),
plot.title = element_text(size = 20, hjust = 0.5),
axis.title = element_blank(),
axis.text.x = element_blank(),
axis.text.y = element_blank(),
axis.ticks = element_blank(),
legend.text = element_text(size = 10),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, paste0("proportion_", celltype, "_STitch3D", ".pdf")),
plot = p2, width = 4, height = 4
)
p1
p2
[24]:
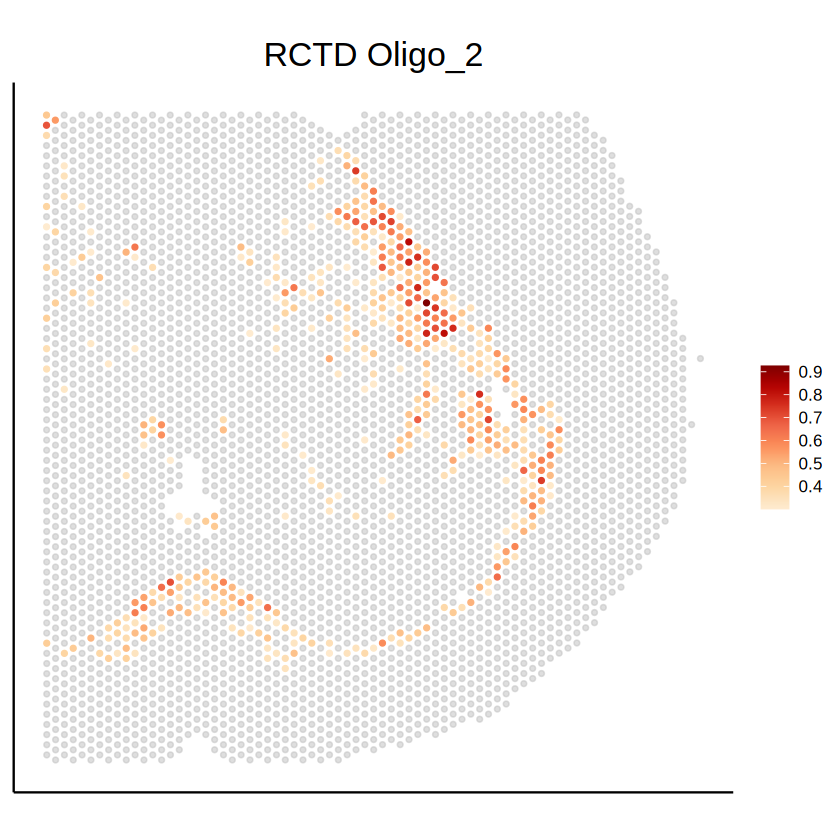
p1 <- mcubePlotPropCellType(
mcube_object_RCTD_1@proportions, mcube_object_RCTD_1@coordinates, celltype,
background = TRUE, proportion_threshold = 0.3
) +
labs(title = paste("RCTD", celltype)) +
theme(
text = element_text(size = 1, hjust = 0.5),
plot.title = element_text(size = 20, hjust = 0.5),
axis.title = element_blank(),
axis.text.x = element_blank(),
axis.text.y = element_blank(),
axis.ticks = element_blank(),
legend.text = element_text(size = 10),
legend.position = "right"
)
ggsave(
filename = file.path(
RESULT_PATH, "visium_1",
paste0("proportion_", celltype, "_region_RCTD", ".pdf")
),
plot = p1, width = 4, height = 4
)
p2 <- mcubePlotPropCellType(
mcube_object_ST_1@proportions, mcube_object_ST_1@coordinates,
celltype, background = TRUE) +
labs(title = paste("STitch3D", celltype)) +
theme(
text = element_text(size = 1, hjust = 0.5),
plot.title = element_text(size = 20, hjust = 0.5),
axis.title = element_blank(),
axis.text.x = element_blank(),
axis.text.y = element_blank(),
axis.ticks = element_blank(),
legend.text = element_text(size = 10),
legend.position = "right"
)
ggsave(
filename = file.path(
RESULT_PATH, "visium_1",
paste0("proportion_", celltype, "_region_STitch3D", ".pdf")
),
plot = p2, width = 4, height = 4
)
p1
p2
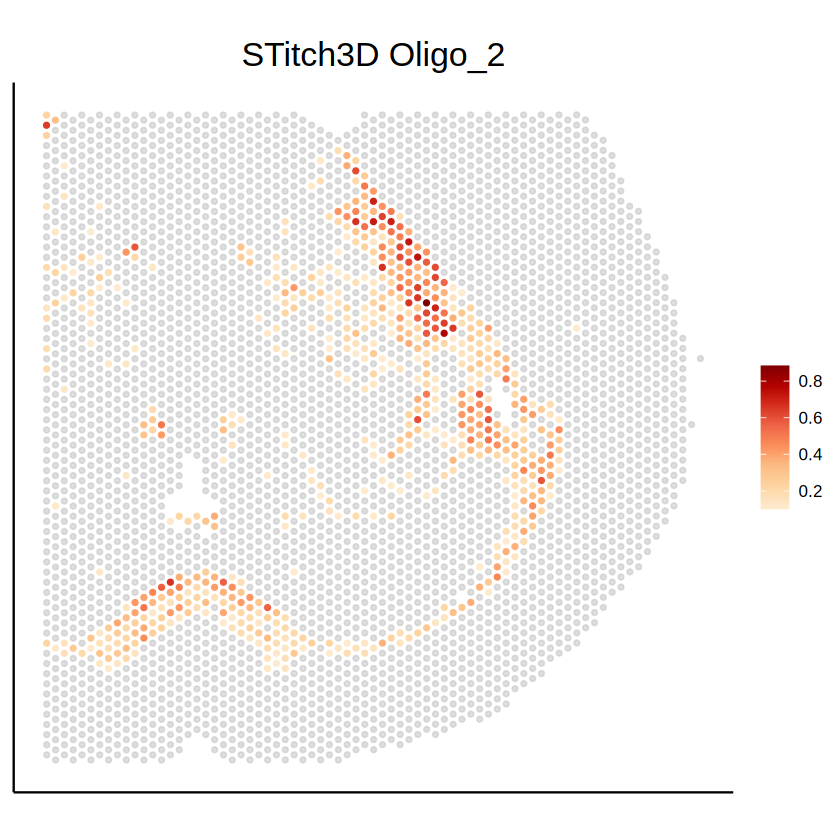
[25]:
mcube_region_RCTD <- createMCube(
counts = t(as.matrix(myRCTD@originalSpatialRNA@counts[, spot_names])),
coordinates = as.matrix(myRCTD@spatialRNA@coords),
proportions = weights_RCTD / rowSums(weights_RCTD),
library_sizes = myRCTD@spatialRNA@nUMI,
reference = t(myRCTD@cell_type_info$info[[1]]),
used_for_deconvolution = rownames(myRCTD@spatialRNA@counts),
spot_effects = spot_effects_RCTD,
celltype_test = "Oligo_2",
celltype_threshold = 50, proportion_threshold = 0.3,
project = "mouse_brain_visium_1"
)
mcube_region_RCTD <- mcubeFitNull(
mcube_region_RCTD,
num_workers = 36, num_threads = 1
)
mcube_region_RCTD <- mcubeTest(
mcube_region_RCTD,
num_workers = 36, num_threads = 1, shared_memory = TRUE
)
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.3 and celltype_threshold = 50.
mcubeFilterCellTypes: Cell type(s) Ext_L23, Oligo_2, Inh_1, Ext_Thal_1, Ext_L56, Ext_Thal_2, Ext_Hpc_DG1, Ext_Hpc_CA1 pass the threshold.
Cell type(s) Oligo_2 will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 2768 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 1347 genes to analyze for Oligo_2.
Preprocessed data description: 4167 spots and 58 cell types in total. 483 spots, 1347 genes, and 1 cell type(s) to analyze.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.101063440730398 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.142925288541018 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.101063440730398 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.142925288541018 for the Gaussian_transformed kernel.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
[26]:
mcube_region_ST <- createMCube(
counts = counts,
coordinates = coordinates,
proportions = proportions_ST,
library_sizes = library_sizes_ST,
reference = reference_ST,
spot_effects = spot_effects_ST,
celltype_test = "Oligo_2",
celltype_threshold = 50,
project = "mouse_brain_visium_1"
)
mcube_region_ST <- mcubeFitNull(
mcube_region_ST,
num_workers = 36, num_threads = 1
)
mcube_region_ST <- mcubeTest(
mcube_region_ST,
num_workers = 36, num_threads = 1, shared_memory = TRUE
)
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.1 and celltype_threshold = 50.
mcubeFilterCellTypes: Cell type(s) Astro_HYPO, Ext_Amy_2, Ext_Hpc_CA1, Ext_Hpc_DG1, Ext_L23, Ext_L25, Ext_L56, Ext_L5_1, Ext_L5_2, Ext_Pir, Ext_Thal_1, Ext_Thal_2, Ext_Unk_3, Inh_1, Inh_2, Inh_3, Inh_4, Oligo_1, Oligo_2 pass the threshold.
Cell type(s) Oligo_2 will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 2599 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 991 genes to analyze for Oligo_2.
Preprocessed data description: 4168 spots and 58 cell types in total. 628 spots, 991 genes, and 1 cell type(s) to analyze.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.101059327818091 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.142919472004653 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.101059327818091 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.142919472004653 for the Gaussian_transformed kernel.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
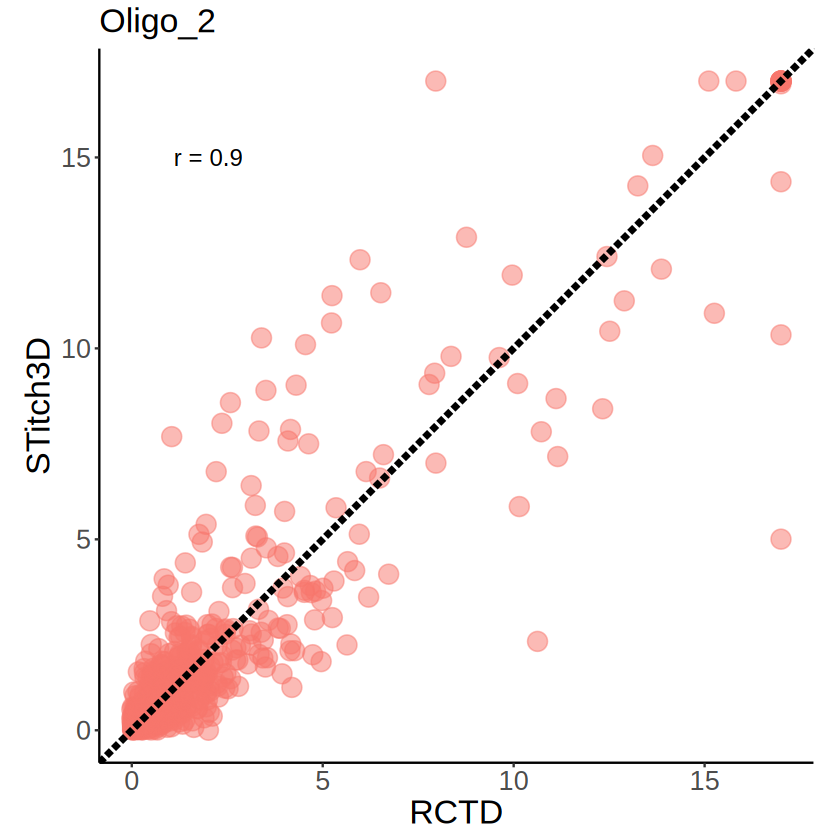
[27]:
mcube_pvalues_region_RCTD <- mcubePvalueList2LongTable(mcube_region_RCTD@pvalues)
mcube_pvalues_region_RCTD$minus_log10_pvalue <- -log10(mcube_pvalues_region_RCTD$pvalue)
mcube_pvalues_region_RCTD$minus_log10_pvalue[mcube_pvalues_region_RCTD$minus_log10_pvalue > 17] <- 17
mcube_pvalues_region_ST <- mcubePvalueList2LongTable(mcube_region_ST@pvalues)
mcube_pvalues_region_ST$minus_log10_pvalue <- -log10(mcube_pvalues_region_ST$pvalue)
mcube_pvalues_region_ST$minus_log10_pvalue[mcube_pvalues_region_ST$minus_log10_pvalue > 17] <- 17
pvalues_merge <- merge(
mcube_pvalues_region_RCTD, mcube_pvalues_region_ST,
by = c("celltype", "gene"),
suffixes = c("_RCTD", "_ST")
)
head(pvalues_merge)
| celltype | gene | pvalue_RCTD | minus_log10_pvalue_RCTD | pvalue_ST | minus_log10_pvalue_ST | |
|---|---|---|---|---|---|---|
| <chr> | <chr> | <dbl> | <dbl> | <dbl> | <dbl> | |
| 1 | Oligo_2 | 1110004F10Rik | 0.01716477 | 1.76536211 | 1.537800e-01 | 0.8131003 |
| 2 | Oligo_2 | 1110051M20Rik | 0.01430794 | 1.84442276 | 3.843169e-02 | 1.4153105 |
| 3 | Oligo_2 | 1700025G04Rik | 0.54033421 | 0.26733754 | 4.166653e-01 | 0.3802126 |
| 4 | Oligo_2 | 4930402H24Rik | 0.01426273 | 1.84579720 | 1.176651e-05 | 4.9293522 |
| 5 | Oligo_2 | 4931406P16Rik | 0.87411995 | 0.05842897 | 5.004367e-01 | 0.3006508 |
| 6 | Oligo_2 | 4933431E20Rik | 0.15644749 | 0.80563139 | 4.845538e-02 | 1.3146580 |
[28]:
correlation <- cor(
pvalues_merge$minus_log10_pvalue_RCTD, pvalues_merge$minus_log10_pvalue_ST,
method = "pearson"
)
[29]:
p <- ggplot(pvalues_merge, aes(x = minus_log10_pvalue_RCTD, y = minus_log10_pvalue_ST)) +
geom_point(aes(color = celltype), size = 5, alpha = 0.5) +
geom_abline(slope = 1, intercept = 0, linetype = "longdash", color = "black", linewidth = 1.5) +
annotate(
"text",
label = paste("r =", round(correlation, 2)),
x = 2, y = 15, size = 5
) +
scale_x_continuous(limits = c(0, 17), oob = scales::oob_squish) +
scale_y_continuous(limits = c(0, 17), oob = scales::oob_squish) +
coord_fixed(ratio = 1) +
labs(
title = celltype,
x = "RCTD", y = "STitch3D"
) +
theme_classic() +
theme(
plot.title = element_text(size = 20),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_text(size = 20),
axis.title.y = element_text(size = 20),
legend.position = "none"
)
ggsave(
file.path(RESULT_PATH, "visium_1", "MMM_Oligo_2_RCTD_vs_STitch3D.pdf"),
plot = p,
width = 4, height = 4
)
p
[30]:
rm(mcube_object_RCTD_1, mcube_object_ST_1)
Visium Slice 2
[31]:
counts <- as.data.frame(readr::read_csv(
file.path(RAW_DATA_PATH, "visium_2", "counts.csv")
))
rownames(counts) <- counts[, 1]
counts[, 1] <- NULL
counts <- as.matrix(counts)
dim(counts)
coordinates <- as.data.frame(readr::read_csv(
file.path(RAW_DATA_PATH, "visium_2", "3D_coordinates.csv")
))
rownames(coordinates) <- coordinates[, 1]
coordinates[, c(1, 4)] <- NULL
coordinates <- as.matrix(coordinates)
dim(coordinates)
puck <- SpatialRNA(as.data.frame(coordinates), t(counts), rowSums(counts))
New names:
• `` -> `...1`
Rows: 3925 Columns: 6416
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): ...1
dbl (6415): 0610040J01Rik, 0610043K17Rik, 1110002J07Rik, 1110004F10Rik, 1110...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
- 3925
- 6415
New names:
• `` -> `...1`
Rows: 3925 Columns: 4
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): ...1
dbl (3): x, y, z
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
- 3925
- 2
Integrating with RCTD
Cell type deconvolution
[32]:
# minor_celltypes <- names(which(table(sc_labels) < 25))
myRCTD <- create.RCTD(
puck, reference,
CELL_MIN_INSTANCE = 20, max_cores = 8
)
myRCTD <- run.RCTD(myRCTD, doublet_mode = "full")
saveRDS(myRCTD, file.path(RESULT_PATH, "visium_2", "myRCTD.rds"))
# myRCTD <- readRDS(file.path(RESULT_PATH, "visium_2", "myRCTD.rds"))
Begin: process_cell_type_info
process_cell_type_info: number of cells in reference: 38849
process_cell_type_info: number of genes in reference: 6415
Ext_L23 Oligo_2 Astro_THAL_med Inh_Sst Inh_6
1244 10000 405 640 467
Ext_L5_3 Inh_Meis2_3 Micro Ext_L5_2 Ext_Pir
127 789 1218 166 954
Astro_AMY Inh_2 Inh_1 Inh_Pvalb Inh_Lamp5
498 839 573 804 331
Ext_L25 Nb_2 Ext_Thal_1 Ext_L56 Ext_Unk_2
922 203 1197 1422 205
Ext_Unk_3 Inh_3 Ext_Amy_2 Ext_Thal_2 Ext_L5_1
416 818 797 671 893
OPC_1 Astro_AMY_CTX Ext_Hpc_DG1 Astro_HYPO Inh_4
1015 279 554 451 1389
Inh_Meis2_1 Ext_Hpc_DG2 Ext_Med Inh_Meis2_2 Ext_Hpc_CA1
363 915 70 602 671
Ext_L6B Astro_CTX Ext_Amy_1 Astro_WM Oligo_1
356 443 519 220 613
Astro_STR Ext_L6 Astro_THAL_lat Nb_1 LowQ_2
81 568 332 225 265
Inh_Vip Astro_HPC Ext_ClauPyr Ext_Hpc_CA2 OPC_2
545 261 272 48 300
Inh_5 Ext_Hpc_CA3 Ext_Unk_1 Inh_Meis2_4 Unk_2
118 232 90 193 85
Astro_THAL_hab LowQ_1 Endo
43 110 22
End: process_cell_type_info
create.RCTD: getting regression differentially expressed genes:
get_de_genes: Ext_L23 found DE genes: 112
get_de_genes: Oligo_2 found DE genes: 408
get_de_genes: Astro_THAL_med found DE genes: 353
get_de_genes: Inh_Sst found DE genes: 123
get_de_genes: Inh_6 found DE genes: 195
get_de_genes: Ext_L5_3 found DE genes: 133
get_de_genes: Inh_Meis2_3 found DE genes: 153
get_de_genes: Micro found DE genes: 647
get_de_genes: Ext_L5_2 found DE genes: 102
get_de_genes: Ext_Pir found DE genes: 69
get_de_genes: Astro_AMY found DE genes: 333
get_de_genes: Inh_2 found DE genes: 99
get_de_genes: Inh_1 found DE genes: 86
get_de_genes: Inh_Pvalb found DE genes: 132
get_de_genes: Inh_Lamp5 found DE genes: 167
get_de_genes: Ext_L25 found DE genes: 147
get_de_genes: Nb_2 found DE genes: 364
get_de_genes: Ext_Thal_1 found DE genes: 212
get_de_genes: Ext_L56 found DE genes: 157
get_de_genes: Ext_Unk_2 found DE genes: 150
get_de_genes: Ext_Unk_3 found DE genes: 150
get_de_genes: Inh_3 found DE genes: 88
get_de_genes: Ext_Amy_2 found DE genes: 74
get_de_genes: Ext_Thal_2 found DE genes: 151
get_de_genes: Ext_L5_1 found DE genes: 105
get_de_genes: OPC_1 found DE genes: 247
get_de_genes: Astro_AMY_CTX found DE genes: 348
get_de_genes: Ext_Hpc_DG1 found DE genes: 196
get_de_genes: Astro_HYPO found DE genes: 352
get_de_genes: Inh_4 found DE genes: 83
get_de_genes: Inh_Meis2_1 found DE genes: 127
get_de_genes: Ext_Hpc_DG2 found DE genes: 184
get_de_genes: Ext_Med found DE genes: 109
get_de_genes: Inh_Meis2_2 found DE genes: 156
get_de_genes: Ext_Hpc_CA1 found DE genes: 185
get_de_genes: Ext_L6B found DE genes: 155
get_de_genes: Astro_CTX found DE genes: 330
get_de_genes: Ext_Amy_1 found DE genes: 112
get_de_genes: Astro_WM found DE genes: 315
get_de_genes: Oligo_1 found DE genes: 355
get_de_genes: Astro_STR found DE genes: 343
get_de_genes: Ext_L6 found DE genes: 95
get_de_genes: Astro_THAL_lat found DE genes: 328
get_de_genes: Nb_1 found DE genes: 239
get_de_genes: LowQ_2 found DE genes: 157
get_de_genes: Inh_Vip found DE genes: 120
get_de_genes: Astro_HPC found DE genes: 337
get_de_genes: Ext_ClauPyr found DE genes: 135
get_de_genes: Ext_Hpc_CA2 found DE genes: 154
get_de_genes: OPC_2 found DE genes: 358
get_de_genes: Inh_5 found DE genes: 172
get_de_genes: Ext_Hpc_CA3 found DE genes: 135
get_de_genes: Ext_Unk_1 found DE genes: 181
get_de_genes: Inh_Meis2_4 found DE genes: 166
get_de_genes: Unk_2 found DE genes: 172
get_de_genes: Astro_THAL_hab found DE genes: 378
get_de_genes: LowQ_1 found DE genes: 416
get_de_genes: Endo found DE genes: 648
get_de_genes: total DE genes: 3432
create.RCTD: getting platform effect normalization differentially expressed genes:
get_de_genes: Ext_L23 found DE genes: 281
get_de_genes: Oligo_2 found DE genes: 585
get_de_genes: Astro_THAL_med found DE genes: 595
get_de_genes: Inh_Sst found DE genes: 234
get_de_genes: Inh_6 found DE genes: 391
get_de_genes: Ext_L5_3 found DE genes: 315
get_de_genes: Inh_Meis2_3 found DE genes: 332
get_de_genes: Micro found DE genes: 969
get_de_genes: Ext_L5_2 found DE genes: 267
get_de_genes: Ext_Pir found DE genes: 214
get_de_genes: Astro_AMY found DE genes: 578
get_de_genes: Inh_2 found DE genes: 276
get_de_genes: Inh_1 found DE genes: 289
get_de_genes: Inh_Pvalb found DE genes: 302
get_de_genes: Inh_Lamp5 found DE genes: 337
get_de_genes: Ext_L25 found DE genes: 327
get_de_genes: Nb_2 found DE genes: 719
get_de_genes: Ext_Thal_1 found DE genes: 402
get_de_genes: Ext_L56 found DE genes: 334
get_de_genes: Ext_Unk_2 found DE genes: 317
get_de_genes: Ext_Unk_3 found DE genes: 315
get_de_genes: Inh_3 found DE genes: 267
get_de_genes: Ext_Amy_2 found DE genes: 225
get_de_genes: Ext_Thal_2 found DE genes: 326
get_de_genes: Ext_L5_1 found DE genes: 275
get_de_genes: OPC_1 found DE genes: 431
get_de_genes: Astro_AMY_CTX found DE genes: 599
get_de_genes: Ext_Hpc_DG1 found DE genes: 376
get_de_genes: Astro_HYPO found DE genes: 591
get_de_genes: Inh_4 found DE genes: 303
get_de_genes: Inh_Meis2_1 found DE genes: 323
get_de_genes: Ext_Hpc_DG2 found DE genes: 361
get_de_genes: Ext_Med found DE genes: 298
get_de_genes: Inh_Meis2_2 found DE genes: 348
get_de_genes: Ext_Hpc_CA1 found DE genes: 323
get_de_genes: Ext_L6B found DE genes: 332
get_de_genes: Astro_CTX found DE genes: 545
get_de_genes: Ext_Amy_1 found DE genes: 295
get_de_genes: Astro_WM found DE genes: 566
get_de_genes: Oligo_1 found DE genes: 521
get_de_genes: Astro_STR found DE genes: 577
get_de_genes: Ext_L6 found DE genes: 286
get_de_genes: Astro_THAL_lat found DE genes: 570
get_de_genes: Nb_1 found DE genes: 523
get_de_genes: LowQ_2 found DE genes: 308
get_de_genes: Inh_Vip found DE genes: 252
get_de_genes: Astro_HPC found DE genes: 572
get_de_genes: Ext_ClauPyr found DE genes: 299
get_de_genes: Ext_Hpc_CA2 found DE genes: 292
get_de_genes: OPC_2 found DE genes: 551
get_de_genes: Inh_5 found DE genes: 389
get_de_genes: Ext_Hpc_CA3 found DE genes: 255
get_de_genes: Ext_Unk_1 found DE genes: 397
get_de_genes: Inh_Meis2_4 found DE genes: 345
get_de_genes: Unk_2 found DE genes: 377
get_de_genes: Astro_THAL_hab found DE genes: 689
get_de_genes: LowQ_1 found DE genes: 811
get_de_genes: Endo found DE genes: 999
get_de_genes: total DE genes: 4489
fitBulk: decomposing bulk
chooseSigma: using initial Q_mat with sigma = 1
Likelihood value: 3477085.10018692
Sigma value: 0.84
Likelihood value: 3407256.54450731
Sigma value: 0.69
Likelihood value: 3350849.20022504
Sigma value: 0.61
Likelihood value: 3326070.00234539
Sigma value: 0.53
Likelihood value: 3306210.96510287
Sigma value: 0.45
Likelihood value: 3292424.52368184
Sigma value: 0.37
Likelihood value: 3286007.90768412
Sigma value: 0.35
Likelihood value: 3285716.09270385
Sigma value: 0.35
[33]:
write.table(
rownames(myRCTD@spatialRNA@counts),
file = file.path(
DATA_PATH, "visium_2",
paste0("HVG_RCTD", ".csv")
),
quote = FALSE, row.names = FALSE, col.names = FALSE
)
[34]:
weights_RCTD <- as.matrix(myRCTD@results$weights)
proportions_RCTD <- weights_RCTD / rowSums(weights_RCTD)
proportions_filter <- proportions_RCTD
proportions_filter[proportions_filter < 0.1] <- 0
major_celltypes <- colnames(proportions_filter)[
which(colSums(proportions_filter) > 50)
]
proportions_RCTD_long_2 <- proportions_RCTD[, major_celltypes] %>%
cbind(myRCTD@spatialRNA@coords) %>%
tidyr::pivot_longer(
cols = -(x:y),
names_to = "Cell type",
values_to = "Proportion"
)
head(proportions_RCTD_long_2)
| x | y | Cell type | Proportion |
|---|---|---|---|
| <dbl> | <dbl> | <chr> | <dbl> |
| 4359.614 | 5109.826 | Ext_L23 | 1.971831e-05 |
| 4359.614 | 5109.826 | Oligo_2 | 7.585819e-02 |
| 4359.614 | 5109.826 | Inh_1 | 1.971831e-05 |
| 4359.614 | 5109.826 | Ext_L25 | 5.911649e-02 |
| 4359.614 | 5109.826 | Ext_Thal_1 | 1.971831e-05 |
| 4359.614 | 5109.826 | Ext_L56 | 1.298476e-01 |
[35]:
p <- ggplot(proportions_RCTD_long_2, aes(x = x, y = y)) +
geom_point(aes(color = Proportion), size = 1, alpha = 1) +
scale_colour_gradientn(name = NULL, colors = pals::brewer.orrd(22)[3:22]) +
scale_y_continuous(trans = "reverse") +
coord_fixed(ratio = 1) +
facet_wrap(~`Cell type`, ncol = 6) +
labs(
title = "Estimated cell type proportions by RCTD",
x = "x", y = "y"
) +
theme_classic() +
theme(
plot.title = element_text(size = 24, hjust = 0.5),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, "visium_2", "proportions_RCTD.pdf"),
plot = p, width = 16, height = 9
)
Identifying cell-type-specific SVGs using MCube
[36]:
spot_names <- colnames(myRCTD@spatialRNA@counts)
weights_RCTD <- as.matrix(myRCTD@results$weights)
spot_effects_RCTD <- log(rowSums(weights_RCTD))
names(spot_effects_RCTD) <- rownames(weights_RCTD)
[37]:
mcube_object_RCTD_2 <- createMCube(
counts = t(as.matrix(myRCTD@originalSpatialRNA@counts[, spot_names])),
coordinates = as.matrix(myRCTD@spatialRNA@coords),
proportions = weights_RCTD / rowSums(weights_RCTD),
library_sizes = myRCTD@spatialRNA@nUMI,
reference = t(myRCTD@cell_type_info$info[[1]]),
used_for_deconvolution = rownames(myRCTD@spatialRNA@counts),
spot_effects = spot_effects_RCTD,
celltype_test = grep("Unk|LowQ", levels(sc_labels), value = TRUE, invert = TRUE),
celltype_threshold = 50,
project = "mouse_brain_visium_2"
)
mcube_object_RCTD_2 <- mcubeFitNull(
mcube_object_RCTD_2,
num_workers = 36, num_threads = 1
)
mcube_object_RCTD_2 <- mcubeTest(
mcube_object_RCTD_2,
num_workers = 36, num_threads = 1, shared_memory = TRUE
)
saveRDS(
mcube_object_RCTD_2,
file = file.path(
RESULT_PATH, "visium_2",
paste0("mcube_RCTD", ".rds")
)
)
saveRDS(
mcube_object_RCTD_2@pvalues,
file = file.path(
RESULT_PATH, "visium_2",
paste0("mcube_pvalues_RCTD", ".rds")
)
)
rm(mcube_object_RCTD_2)
# mcube_pvalues_RCTD_2 <- readRDS(
# file = file.path(
# RESULT_PATH, "visium_2",
# paste0("mcube_pvalues_RCTD", ".rds")
# )
# )
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.1 and celltype_threshold = 50.
mcubeFilterCellTypes: Cell type(s) Ext_L23, Oligo_2, Inh_1, Ext_L25, Ext_Thal_1, Ext_L56, Ext_Thal_2, Ext_L5_1, Ext_Hpc_DG1, Astro_HYPO, Inh_4, Ext_Med, Ext_Hpc_CA1, Oligo_1, Astro_HPC pass the threshold.
Cell type(s) Astro_THAL_med, Inh_Sst, Inh_6, Ext_L5_3, Inh_Meis2_3, Micro, Ext_L5_2, Ext_Pir, Astro_AMY, Inh_2, Inh_Pvalb, Inh_Lamp5, Nb_2, Inh_3, Ext_Amy_2, OPC_1, Astro_AMY_CTX, Inh_Meis2_1, Ext_Hpc_DG2, Inh_Meis2_2, Ext_L6B, Astro_CTX, Ext_Amy_1, Astro_WM, Astro_STR, Ext_L6, Astro_THAL_lat, Nb_1, Inh_Vip, Ext_ClauPyr, Ext_Hpc_CA2, OPC_2, Inh_5, Ext_Hpc_CA3, Inh_Meis2_4, Astro_THAL_hab, Endo has/have less than the minimum celltype_threshold = 50 with proportion_threshold = 0.1.
To include the above cell type(s), please reduce celltype_threshold or proportion_threshold.
Cell type(s) Ext_L23, Oligo_2, Inh_1, Ext_L25, Ext_Thal_1, Ext_L56, Ext_Thal_2, Ext_L5_1, Ext_Hpc_DG1, Astro_HYPO, Inh_4, Ext_Med, Ext_Hpc_CA1, Oligo_1, Astro_HPC will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 2767 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 954 genes to analyze for Ext_L23.
mcubeFilterGenesCellType: Select 1073 genes to analyze for Oligo_2.
mcubeFilterGenesCellType: Select 1161 genes to analyze for Inh_1.
mcubeFilterGenesCellType: Select 989 genes to analyze for Ext_L25.
mcubeFilterGenesCellType: Select 938 genes to analyze for Ext_Thal_1.
mcubeFilterGenesCellType: Select 1024 genes to analyze for Ext_L56.
mcubeFilterGenesCellType: Select 1041 genes to analyze for Ext_Thal_2.
mcubeFilterGenesCellType: Select 967 genes to analyze for Ext_L5_1.
mcubeFilterGenesCellType: Select 981 genes to analyze for Ext_Hpc_DG1.
mcubeFilterGenesCellType: Select 1137 genes to analyze for Astro_HYPO.
mcubeFilterGenesCellType: Select 1162 genes to analyze for Inh_4.
mcubeFilterGenesCellType: Select 1033 genes to analyze for Ext_Med.
mcubeFilterGenesCellType: Select 887 genes to analyze for Ext_Hpc_CA1.
mcubeFilterGenesCellType: Select 869 genes to analyze for Oligo_1.
mcubeFilterGenesCellType: Select 1112 genes to analyze for Astro_HPC.
Preprocessed data description: 3924 spots and 58 cell types in total. 3658 spots, 2756 genes, and 15 cell type(s) to analyze.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.104574506874699 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.147890685900678 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.104574506874699 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.147890685900678 for the Gaussian_transformed kernel.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
Integrating with STitch3D
Load in deconvolution results
[38]:
reference_ST <- data.matrix(read.csv(
file.path(DATA_PATH, "visium_2", "reference_ST.csv"),
header = TRUE, row.names = 1, check.names = FALSE
))
dim(reference_ST)
num_celltypes <- nrow(reference_ST)
proportions_ST <- data.matrix(read.csv(
file.path(DATA_PATH, "visium_2", paste0("prop_slice", 0, ".csv")),
header = TRUE, row.names = 1, check.names = FALSE
))
dim(proportions_ST)
counts <- as.data.frame(readr::read_csv(
file.path(DATA_PATH, "visium_2", "counts.csv")
))
rownames(counts) <- counts[, 1]
counts[, 1] <- NULL
counts <- data.matrix(counts)
dim(counts)
coordinates <- data.matrix(read.csv(
file.path(DATA_PATH, "visium_2", "3D_coordinates.csv"),
header = TRUE, row.names = 1, check.names = FALSE
))[, c("x", "y")]
dim(coordinates)
spot_effects_ST <- data.matrix(read.csv(
file.path(DATA_PATH, "visium_2", "spot_effects_ST.csv"),
header = TRUE, row.names = 1, check.names = FALSE
))[, 1]
library_sizes_ST <- data.matrix(read.csv(
file.path(DATA_PATH, "visium_2", "library_sizes_ST.csv"),
header = TRUE, row.names = 1, check.names = FALSE
))[, 1]
- 58
- 4489
- 3925
- 58
New names:
• `` -> `...1`
Rows: 3925 Columns: 4490
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (1): ...1
dbl (4489): Xkr4, Sox17, Rgs20, Oprk1, Rb1cc1, St18, Sntg1, Adhfe1, Sgk3, Mc...
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
- 3925
- 4489
- 3925
- 2
[39]:
proportions_filter <- proportions_ST
proportions_filter[proportions_filter < 0.1] <- 0
major_celltypes <- colnames(proportions_filter)[
which(colSums(proportions_filter) > 50)
]
proportions_ST_long_2 <- as.data.frame(proportions_ST[, major_celltypes]) %>%
cbind(coordinates) %>%
tidyr::pivot_longer(
cols = -(x:y),
names_to = "Cell type",
values_to = "Proportion"
)
head(proportions_ST_long_2)
| x | y | Cell type | Proportion |
|---|---|---|---|
| <dbl> | <dbl> | <chr> | <dbl> |
| 4359.614 | 5109.826 | Astro_HYPO | 1.179485e-06 |
| 4359.614 | 5109.826 | Ext_Amy_2 | 7.797921e-05 |
| 4359.614 | 5109.826 | Ext_Hpc_CA1 | 1.753935e-04 |
| 4359.614 | 5109.826 | Ext_Hpc_DG1 | 1.280550e-07 |
| 4359.614 | 5109.826 | Ext_L23 | 1.618132e-04 |
| 4359.614 | 5109.826 | Ext_L25 | 6.004004e-02 |
[40]:
p <- ggplot(proportions_ST_long_2, aes(x = x, y = y)) +
geom_point(aes(color = Proportion), size = 1, alpha = 1) +
scale_colour_gradientn(name = NULL, colors = pals::brewer.orrd(22)[3:22]) +
scale_y_continuous(trans = "reverse") +
coord_fixed(ratio = 1) +
facet_wrap(~`Cell type`, nrow = 3) +
labs(
title = "Estimated cell type proportions by STitch3D",
x = "x", y = "y"
) +
theme_classic() +
theme(
plot.title = element_text(size = 24, hjust = 0.5),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, "visium_2", "proportions_STitch3D.pdf"),
plot = p, width = 16, height = 9
)
Identifying cell-type-specific SVGs using MCube
[41]:
mcube_object_ST_2 <- createMCube(
counts = counts,
coordinates = coordinates,
proportions = proportions_ST,
library_sizes = library_sizes_ST,
reference = reference_ST,
spot_effects = spot_effects_ST,
celltype_test = grep("Unk|LowQ", levels(sc_labels), value = TRUE, invert = TRUE),
celltype_threshold = 50,
project = "mouse_brain_visium_2"
)
mcube_object_ST_2 <- mcubeFitNull(
mcube_object_ST_2,
num_workers = 36, num_threads = 1
)
mcube_object_ST_2 <- mcubeTest(
mcube_object_ST_2,
num_workers = 36, num_threads = 1, shared_memory = TRUE
)
saveRDS(
mcube_object_ST_2,
file = file.path(
RESULT_PATH, "visium_2",
paste0("mcube_ST", ".rds")
)
)
saveRDS(
mcube_object_ST_2@pvalues,
file = file.path(
RESULT_PATH, "visium_2",
paste0("mcube_pvalues_ST", ".rds")
)
)
rm(mcube_object_ST_2)
# mcube_pvalues_ST_2 <- readRDS(
# file = file.path(
# RESULT_PATH, "sensitivity", "visium_2",
# paste0("mcube_pvalues_ST", ".rds")
# )
# )
The batch_id is not provided!
All spots are assumed to be from the same batch and share the same gene platform effects.
Select high-abundance cell types to analyze with proportion_threshold = 0.1 and celltype_threshold = 50.
mcubeFilterCellTypes: Cell type(s) Astro_HYPO, Ext_Amy_2, Ext_Hpc_CA1, Ext_Hpc_DG1, Ext_L23, Ext_L25, Ext_L56, Ext_L5_1, Ext_L5_2, Ext_Med, Ext_Thal_1, Ext_Thal_2, Ext_Unk_3, Inh_1, Inh_2, Inh_3, Inh_4, Oligo_1, Oligo_2 pass the threshold.
Cell type(s) Astro_AMY, Astro_AMY_CTX, Astro_CTX, Astro_HPC, Astro_STR, Astro_THAL_hab, Astro_THAL_lat, Astro_THAL_med, Astro_WM, Endo, Ext_Amy_1, Ext_ClauPyr, Ext_Hpc_CA2, Ext_Hpc_CA3, Ext_Hpc_DG2, Ext_L5_3, Ext_L6, Ext_L6B, Ext_Pir, Inh_5, Inh_6, Inh_Lamp5, Inh_Meis2_1, Inh_Meis2_2, Inh_Meis2_3, Inh_Meis2_4, Inh_Pvalb, Inh_Sst, Inh_Vip, Micro, Nb_1, Nb_2, OPC_1, OPC_2 has/have less than the minimum celltype_threshold = 50 with proportion_threshold = 0.1.
To include the above cell type(s), please reduce celltype_threshold or proportion_threshold.
Cell type(s) Astro_HYPO, Ext_Amy_2, Ext_Hpc_CA1, Ext_Hpc_DG1, Ext_L23, Ext_L25, Ext_L56, Ext_L5_1, Ext_L5_2, Ext_Med, Ext_Thal_1, Ext_Thal_2, Inh_1, Inh_2, Inh_3, Inh_4, Oligo_1, Oligo_2 will be analyzed.
Filter out lowly-expressed genes with gene_threshold = 5e-05.
mcubeFilterGenes: 2602 genes pass the threshold.
The platform effects are not provided and need to be estimated from data!
Select highly-expressed genes to analyze for each specific cell type with reference_threshold = 0.5.
mcubeFilterGenesCellType: Select 1074 genes to analyze for Astro_HYPO.
mcubeFilterGenesCellType: Select 917 genes to analyze for Ext_Amy_2.
mcubeFilterGenesCellType: Select 802 genes to analyze for Ext_Hpc_CA1.
mcubeFilterGenesCellType: Select 906 genes to analyze for Ext_Hpc_DG1.
mcubeFilterGenesCellType: Select 870 genes to analyze for Ext_L23.
mcubeFilterGenesCellType: Select 890 genes to analyze for Ext_L25.
mcubeFilterGenesCellType: Select 944 genes to analyze for Ext_L56.
mcubeFilterGenesCellType: Select 884 genes to analyze for Ext_L5_1.
mcubeFilterGenesCellType: Select 848 genes to analyze for Ext_L5_2.
mcubeFilterGenesCellType: Select 938 genes to analyze for Ext_Med.
mcubeFilterGenesCellType: Select 876 genes to analyze for Ext_Thal_1.
mcubeFilterGenesCellType: Select 961 genes to analyze for Ext_Thal_2.
mcubeFilterGenesCellType: Select 1079 genes to analyze for Inh_1.
mcubeFilterGenesCellType: Select 1001 genes to analyze for Inh_2.
mcubeFilterGenesCellType: Select 1069 genes to analyze for Inh_3.
mcubeFilterGenesCellType: Select 1077 genes to analyze for Inh_4.
mcubeFilterGenesCellType: Select 799 genes to analyze for Oligo_1.
mcubeFilterGenesCellType: Select 985 genes to analyze for Oligo_2.
Preprocessed data description: 3925 spots and 58 cell types in total. 3658 spots, 2586 genes, and 18 cell type(s) to analyze.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
mcubeKernel: length_scale is set as 0.104577495815249 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.147894912900941 for the Gaussian kernel.
mcubeKernel: length_scale is set as 0.104577495815249 for the Gaussian_transformed kernel.
mcubeKernel: length_scale is set as 0.147894912900941 for the Gaussian_transformed kernel.
Number of physical cores: 40.
Number of workers: 36.
Number of thread(s) on BLAS per worker: 1.
Comparison of the deconvolution results
[42]:
overlap_celltypes <- intersect(
unique(proportions_RCTD_long_2$`Cell type`),
unique(proportions_ST_long_2$`Cell type`)
)
overlap_celltypes <- sort(overlap_celltypes)
overlap_celltypes
- 'Astro_HYPO'
- 'Ext_Hpc_CA1'
- 'Ext_Hpc_DG1'
- 'Ext_L23'
- 'Ext_L25'
- 'Ext_L5_1'
- 'Ext_L56'
- 'Ext_Med'
- 'Ext_Thal_1'
- 'Ext_Thal_2'
- 'Inh_1'
- 'Inh_4'
- 'Oligo_1'
- 'Oligo_2'
[43]:
p <- ggplot(
proportions_RCTD_long_2[proportions_RCTD_long_2$`Cell type` %in% overlap_celltypes, ],
aes(x = x, y = y)
) +
geom_point(aes(color = Proportion), size = 1, alpha = 1) +
scale_colour_gradientn(name = NULL, colors = pals::brewer.orrd(22)[3:22]) +
scale_y_continuous(trans = "reverse") +
coord_fixed(ratio = 1) +
facet_wrap(~`Cell type`, nrow = 2) +
labs(
title = "Estimated cell type proportions by RCTD",
x = "x", y = "y"
) +
theme_classic() +
theme(
plot.title = element_text(size = 24, hjust = 0.5),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, "visium_2", "sensitivity_proportions_RCTD.pdf"),
plot = p, width = 16, height = 6
)
[44]:
p <- ggplot(
proportions_ST_long_2[proportions_ST_long_2$`Cell type` %in% overlap_celltypes, ],
aes(x = x, y = y)
) +
geom_point(aes(color = Proportion), size = 1, alpha = 1) +
scale_colour_gradientn(name = NULL, colors = pals::brewer.orrd(22)[3:22]) +
scale_y_continuous(trans = "reverse") +
coord_fixed(ratio = 1) +
facet_wrap(~`Cell type`, nrow = 2) +
labs(
title = "Estimated cell type proportions by STitch3D",
x = "x", y = "y"
) +
theme_classic() +
theme(
plot.title = element_text(size = 24, hjust = 0.5),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, "visium_2", "sensitivity_proportions_STitch3D.pdf"),
plot = p, width = 16, height = 6
)
Comparison of the cell-type-specific SVG results
[45]:
mcube_pvalues_RCTD_2 <- readRDS(
file = file.path(
RESULT_PATH, "visium_2",
paste0("mcube_pvalues_RCTD", ".rds")
)
)
mcube_pvalues_ST_2 <- readRDS(
file = file.path(
RESULT_PATH, "visium_2",
paste0("mcube_pvalues_ST", ".rds")
)
)
[46]:
mcube_pvalues_RCTD_2 <- mcubePvalueList2LongTable(mcube_pvalues_RCTD_2)
mcube_pvalues_RCTD_2$minus_log10_pvalue <- -log10(mcube_pvalues_RCTD_2$pvalue)
mcube_pvalues_RCTD_2$minus_log10_pvalue[mcube_pvalues_RCTD_2$minus_log10_pvalue > 17] <- 17
mcube_pvalues_ST_2 <- mcubePvalueList2LongTable(mcube_pvalues_ST_2)
mcube_pvalues_ST_2$minus_log10_pvalue <- -log10(mcube_pvalues_ST_2$pvalue)
mcube_pvalues_ST_2$minus_log10_pvalue[mcube_pvalues_ST_2$minus_log10_pvalue > 17] <- 17
pvalues_merge <- merge(
mcube_pvalues_RCTD_2, mcube_pvalues_ST_2,
by = c("celltype", "gene"),
suffixes = c("_RCTD", "_ST")
)
head(pvalues_merge)
| celltype | gene | pvalue_RCTD | minus_log10_pvalue_RCTD | pvalue_ST | minus_log10_pvalue_ST | |
|---|---|---|---|---|---|---|
| <chr> | <chr> | <dbl> | <dbl> | <dbl> | <dbl> | |
| 1 | Astro_HYPO | 1110004F10Rik | 0.001278629 | 2.89325554 | 0.03689844 | 1.4329920 |
| 2 | Astro_HYPO | 1110051M20Rik | 0.010943041 | 1.96086199 | 0.07129424 | 1.1469455 |
| 3 | Astro_HYPO | 2310022B05Rik | 0.902371928 | 0.04461442 | 0.33203934 | 0.4788105 |
| 4 | Astro_HYPO | 2700081O15Rik | 0.157050125 | 0.80396171 | 0.31020245 | 0.5083548 |
| 5 | Astro_HYPO | 4930402H24Rik | 0.716225347 | 0.14495031 | 0.38755751 | 0.4116638 |
| 6 | Astro_HYPO | 4933431E20Rik | 0.617446854 | 0.20940042 | 0.37551396 | 0.4253739 |
[47]:
correlations <- pvalues_merge %>%
group_by(celltype) %>%
summarize(
correlation = cor(minus_log10_pvalue_RCTD, minus_log10_pvalue_ST, method = "pearson")
)
[48]:
p <- ggplot(pvalues_merge, aes(x = -log10(pvalue_RCTD), y = -log10(pvalue_ST))) +
geom_point(aes(color = celltype), size = 5, alpha = 0.5) +
geom_abline(slope = 1, intercept = 0, linetype = "longdash", color = "black", linewidth = 1.5) +
geom_text(
data = correlations,
mapping = aes(
x = 3, y = 15,
label = paste("r =", round(correlation, 2))
),
size = 5
) +
scale_x_continuous(limits = c(0, 17), oob = scales::oob_squish) +
scale_y_continuous(limits = c(0, 17), oob = scales::oob_squish) +
coord_fixed(ratio = 1) +
labs(
title = expression(paste("Comparison of ", "-log"[10], plain("P"), " from MMM")),
x = "RCTD", y = "STitch3D"
) +
facet_wrap(celltype ~ ., nrow = 2) +
theme_classic() +
theme(
plot.title = element_text(size = 24),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_text(size = 20),
axis.title.y = element_text(size = 20),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "none"
)
ggsave(
file.path(RESULT_PATH, "visium_2", "MMM_RCTD_vs_STitch3D.pdf"),
plot = p,
width = 15, height = 6
)
ggsave(
file.path(RESULT_PATH, "visium_2", "MMM_RCTD_vs_STitch3D.png"),
plot = p,
width = 15, height = 6
)
p
Replicability analysis of MCube using the two slices
Comparison of the deconvolution results from RCTD on the two slices
[49]:
overlap_celltypes <- intersect(
unique(proportions_RCTD_long_1$`Cell type`),
unique(proportions_RCTD_long_2$`Cell type`)
)
overlap_celltypes <- sort(overlap_celltypes)
overlap_celltypes
- 'Astro_HPC'
- 'Astro_HYPO'
- 'Ext_Hpc_CA1'
- 'Ext_Hpc_DG1'
- 'Ext_L23'
- 'Ext_L25'
- 'Ext_L5_1'
- 'Ext_L56'
- 'Ext_Med'
- 'Ext_Thal_1'
- 'Ext_Thal_2'
- 'Inh_1'
- 'Inh_4'
- 'Oligo_1'
- 'Oligo_2'
[50]:
p <- ggplot(
proportions_RCTD_long_1[proportions_RCTD_long_1$`Cell type` %in% overlap_celltypes, ],
aes(x = x, y = y)
) +
geom_point(aes(color = Proportion), size = 1, alpha = 1) +
scale_colour_gradientn(name = NULL, colors = pals::brewer.orrd(22)[3:22]) +
scale_y_continuous(trans = "reverse") +
coord_fixed(ratio = 1) +
facet_wrap(~`Cell type`, nrow = 2) +
labs(
title = "Estimated cell type proportions by RCTD on Slice 1",
x = "x", y = "y"
) +
theme_classic() +
theme(
plot.title = element_text(size = 24, hjust = 0.5),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, "visium_1", "replicability_proportions_RCTD.pdf"),
plot = p, width = 20, height = 6
)
[51]:
p <- ggplot(
proportions_RCTD_long_2[proportions_RCTD_long_2$`Cell type` %in% overlap_celltypes, ],
aes(x = x, y = y)
) +
geom_point(aes(color = Proportion), size = 1, alpha = 1) +
scale_colour_gradientn(name = NULL, colors = pals::brewer.orrd(22)[3:22]) +
scale_y_continuous(trans = "reverse") +
coord_fixed(ratio = 1) +
facet_wrap(~`Cell type`, nrow = 2) +
labs(
title = "Estimated cell type proportions by RCTD on Slice 2",
x = "x", y = "y"
) +
theme_classic() +
theme(
plot.title = element_text(size = 24, hjust = 0.5),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "right"
)
ggsave(
filename = file.path(RESULT_PATH, "visium_2", "replicability_proportions_RCTD.pdf"),
plot = p, width = 20, height = 6
)
Comparison of the cell-type-specific SVG results on the two slices
[52]:
pvalues_merge <- merge(
mcube_pvalues_RCTD_1, mcube_pvalues_RCTD_2,
by = c("celltype", "gene"),
suffixes = c("_slice1", "_slice2")
)
head(pvalues_merge)
| celltype | gene | pvalue_slice1 | minus_log10_pvalue_slice1 | pvalue_slice2 | minus_log10_pvalue_slice2 | |
|---|---|---|---|---|---|---|
| <chr> | <chr> | <dbl> | <dbl> | <dbl> | <dbl> | |
| 1 | Astro_HPC | 1110004F10Rik | 0.16333938 | 0.7869091 | 0.33869033 | 0.47019720 |
| 2 | Astro_HPC | 1110051M20Rik | 0.11299529 | 0.9469396 | 0.09168947 | 1.03768056 |
| 3 | Astro_HPC | 2700081O15Rik | 0.02374883 | 1.6243577 | 0.07037821 | 1.15256181 |
| 4 | Astro_HPC | 4930402H24Rik | 0.60270832 | 0.2198928 | 0.87571766 | 0.05763589 |
| 5 | Astro_HPC | 4932438A13Rik | 0.17432673 | 0.7586360 | 0.40611553 | 0.39135040 |
| 6 | Astro_HPC | 4933431E20Rik | 0.63832126 | 0.1949607 | 0.47503713 | 0.32327244 |
[53]:
correlations <- pvalues_merge %>%
group_by(celltype) %>%
summarize(
correlation = cor(minus_log10_pvalue_slice1, minus_log10_pvalue_slice2, method = "pearson")
)
[54]:
p <- ggplot(pvalues_merge, aes(x = minus_log10_pvalue_slice1, y = minus_log10_pvalue_slice2)) +
geom_point(aes(color = celltype), size = 5, alpha = 0.5) +
geom_abline(slope = 1, intercept = 0, linetype = "longdash", color = "black", linewidth = 1.5) +
geom_text(
data = correlations,
mapping = aes(
x = 4, y = 15,
label = paste("r =", round(correlation, 2))
),
size = 5
) +
scale_x_continuous(limits = c(0, 17), oob = scales::oob_squish) +
scale_y_continuous(limits = c(0, 17), oob = scales::oob_squish) +
coord_fixed(ratio = 1) +
labs(
title = expression(paste("Comparison of ", "-log"[10], plain("P"), " from MMM on two slices")),
x = "Slice 1", y = "Slice 2"
) +
facet_wrap(celltype ~ ., nrow = 2) +
theme_classic() +
theme(
plot.title = element_text(size = 24),
axis.text.x = element_text(size = 16),
axis.text.y = element_text(size = 16),
axis.title.x = element_text(size = 20),
axis.title.y = element_text(size = 20),
strip.text = element_text(size = 16),
legend.title = element_blank(),
legend.text = element_text(size = 16),
legend.position = "none"
)
ggsave(
file.path(RESULT_PATH, "mouse_brain_MMM_replicability.pdf"),
plot = p,
width = 15, height = 6
)
p
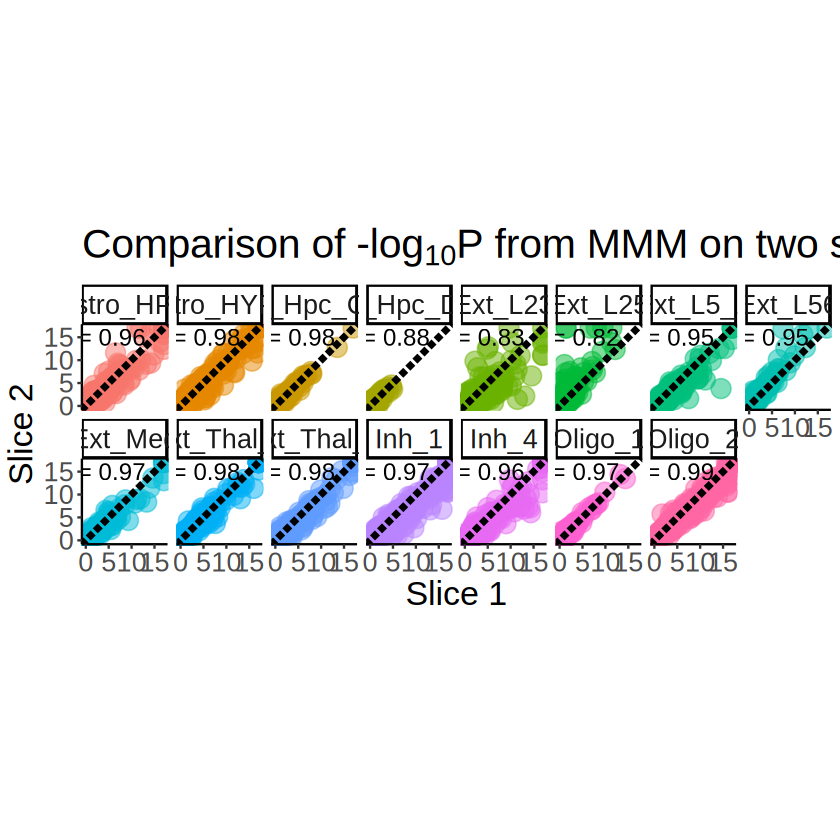
[ ]: