Niche-differential expression analysis using nicheDE
[1]:
set.seed(20240709)
library(spacexr)
library(nicheDE)
library(MCube)
library(ggplot2)
[2]:
DATA_PATH <- "/import/home/share/zw/pql/data/breast_cancer"
RESULT_PATH <- "/import/home/share/zw/pql/results/breast_cancer"
if (!dir.exists(RESULT_PATH)) {
dir.create(RESULT_PATH, recursive = TRUE)
}
Applying RCTD and nicheDE to the ST dataset segmented by UCS
[3]:
seg_method <- "UCS_10X"
myRCTD <- readRDS(file.path(RESULT_PATH, seg_method, "myRCTD.rds"))
[4]:
# read in data
# create library matrix
librarymatrix <- CreateLibraryMatrix(
t(as.matrix(myRCTD@reference@counts)),
cbind(
colnames(myRCTD@reference@counts),
as.character(myRCTD@reference@cell_types)
)
)
[1] "Computing average expression profile matrix"
[1] "Average expression matrix computed"
[5]:
weights_RCTD <- as.matrix(myRCTD@results$weights)
NDE_obj <- CreateNicheDEObject(
t(as.matrix(myRCTD@spatialRNA@counts)), myRCTD@spatialRNA@coords,
librarymatrix, weights_RCTD / rowSums(weights_RCTD),
sigma = c(50, 100, 200)
)
[1] "Creating Niche-DE object"
[1] "Niche-DE object created with 17201 observations, 287 genes, 1 batch(es), and 17 cell types."
[6]:
NDE_obj <- CalculateEffectiveNiche(NDE_obj)
[1] "Calculating effective niche for kernel bandwith 50(1 out of 3 values)."
[1] "Calculating effective niche for kernel bandwith 100(2 out of 3 values)."
[1] "Calculating effective niche for kernel bandwith 200(3 out of 3 values)."
[1] "Effective niche calculated"
[7]:
NDE_obj <- niche_DE(NDE_obj, num_cores = 36, outfile = "")
[1] "Starting Niche-DE analysis with parameters C = 150, M = 10, gamma = 0.8."
[1] "Performing Niche-DE analysis with kernel bandwidth:50 (number 1 out of 3 values)"
[1] "Running Niche-DE in parallel"
[1] "Splitting Data into 1 chunks in order to avoid memory overload. Each chunk is less than 1 gigabytes."
[1] "Initializing cluster"
[1] "Evaluating chunk 1 out of 1"
Warning message in e$fun(obj, substitute(ex), parent.frame(), e$data):
“already exporting variable(s): constant_param, niche_DE_core, counts_chunk”
[1] "Closing cluster"
[1] "Cleaning disk for next iteration"
[1] "Performing Niche-DE analysis with kernel bandwidth:100 (number 2 out of 3 values)"
[1] "Running Niche-DE in parallel"
[1] "Splitting Data into 1 chunks in order to avoid memory overload. Each chunk is less than 1 gigabytes."
[1] "Initializing cluster"
[1] "Evaluating chunk 1 out of 1"
Warning message in e$fun(obj, substitute(ex), parent.frame(), e$data):
“already exporting variable(s): constant_param, niche_DE_core, counts_chunk”
[1] "Closing cluster"
[1] "Cleaning disk for next iteration"
[1] "Performing Niche-DE analysis with kernel bandwidth:200 (number 3 out of 3 values)"
[1] "Running Niche-DE in parallel"
[1] "Splitting Data into 1 chunks in order to avoid memory overload. Each chunk is less than 1 gigabytes."
[1] "Initializing cluster"
[1] "Evaluating chunk 1 out of 1"
Warning message in e$fun(obj, substitute(ex), parent.frame(), e$data):
“already exporting variable(s): constant_param, niche_DE_core, counts_chunk”
[1] "Closing cluster"
[1] "Cleaning disk for next iteration"
[1] "Computing Positive Niche-DE Pvalues"
[1] "Computing Gene Level Pvalues"
[1] "Combining Gene Level Pvalues Across Kernel Bandwidths"
[1] "Computing Cell Type Level Pvalues"
[1] "Combining Cell Type Level Pvalues Across Kernel Bandwidths"
[1] "Computing and Combining interaction Level Pvalues Across Kernel bandwidths"
[1] "Computing Negative Niche-DE Pvalues"
[1] "Computing Gene Level Pvalues"
[1] "Combining Gene Level Pvalues Across Kernel Bandwidths"
[1] "Computing Cell Type Level Pvalues"
[1] "Combining Cell Type Level Pvalues Across Kernel Bandwidths"
[1] "Computing and Combining interaction Level Pvalues Across Kernel bandwidths"
[1] "Niche-DE analysis complete. Number of Genes with niche-DE T-stat equal to 227"
Warning message in niche_DE(NDE_obj, num_cores = 36, outfile = ""):
“Less than 1000 genes pass. This could be due to insufficient read depth of data or size of C parameter. Consider changing choice of C parameter”
[8]:
saveRDS(
object = NDE_obj,
file = file.path(
RESULT_PATH, seg_method,
paste0(
"NDE_obj",
".rds"
)
)
)
# NDE_obj <- readRDS(
# file = file.path(
# RESULT_PATH, seg_method,
# paste0(
# "NDE_obj",
# ".rds"
# )
# )
# )
[9]:
# Target cell type: DCIS 1
# Niche cell type: Invasive Tumor
get_niche_DE_genes(
NDE_obj, test.level = "I",
index = "DCIS 1", niche = "Invasive Tumor",
pos = TRUE, alpha = 0.05
)
get_niche_DE_genes(
NDE_obj, test.level = "I",
index = "DCIS 1", niche = "Invasive Tumor",
pos = FALSE, alpha = 0.05
)
[1] "Returning Niche-DE Genes"
| Genes | Pvalues.Interaction | |
|---|---|---|
| <chr> | <dbl> | |
| 6 | KLF5 | 0.000000e+00 |
| 7 | CDC42EP1 | 0.000000e+00 |
| 10 | SERHL2 | 0.000000e+00 |
| 2 | ACTA2 | 1.421085e-14 |
| 8 | KRT7 | 1.746159e-12 |
| 11 | FSTL3 | 7.120960e-08 |
| 16 | TACSTD2 | 3.502333e-07 |
| 17 | BACE2 | 1.428245e-03 |
| 12 | C6orf132 | 4.869276e-03 |
| 9 | MYO5B | 5.190509e-03 |
| 3 | MZB1 | 9.895541e-03 |
| 1 | USP53 | 1.268764e-02 |
| 14 | TPD52 | 1.391316e-02 |
| 4 | SEC11C | 3.428432e-02 |
| 15 | SVIL | 3.810915e-02 |
| 5 | OCIAD2 | 4.262528e-02 |
| 13 | KRT8 | 4.418016e-02 |
[1] "Returning Niche-DE Genes"
| Genes | Pvalues.Interaction | |
|---|---|---|
| <chr> | <dbl> | |
| 4 | SERPINA3 | 0.000000000 |
| 2 | RHOH | 0.001215106 |
| 3 | FLNB | 0.015552298 |
| 1 | MLPH | 0.016036460 |
| 5 | ANKRD30A | 0.024360175 |
[10]:
# Target cell type: DCIS 2
# Niche cell type: Invasive Tumor
get_niche_DE_genes(
NDE_obj, test.level = "I",
index = "DCIS 2", niche = "Invasive Tumor",
pos = TRUE, alpha = 0.05
)
get_niche_DE_genes(
NDE_obj, test.level = "I",
index = "DCIS 2", niche = "Invasive Tumor",
pos = FALSE, alpha = 0.05
)
[1] "Returning Niche-DE Genes"
| Genes | Pvalues.Interaction | |
|---|---|---|
| <chr> | <dbl> | |
| 8 | SERHL2 | 0.000000e+00 |
| 10 | ERN1 | 0.000000e+00 |
| 4 | AGR3 | 1.184238e-15 |
| 2 | SCD | 2.482522e-08 |
| 12 | LUM | 5.016727e-07 |
| 3 | S100A14 | 1.677298e-06 |
| 13 | TENT5C | 3.242025e-05 |
| 9 | SERPINA3 | 6.913058e-05 |
| 15 | TOMM7 | 7.185086e-04 |
| 5 | ESR1 | 7.284165e-04 |
| 6 | WARS | 5.464156e-03 |
| 11 | CEACAM6 | 5.550267e-03 |
| 16 | FBLN1 | 7.937503e-03 |
| 1 | CAVIN2 | 1.007103e-02 |
| 14 | KRT8 | 2.483159e-02 |
| 7 | FASN | 3.017706e-02 |
[1] "Returning Niche-DE Genes"
| Genes | Pvalues.Interaction | |
|---|---|---|
| <chr> | <dbl> | |
| 1 | TCIM | 1.897227e-09 |
| 2 | KRT7 | 1.374679e-02 |
Comparing niche-associated genes with cell-type-specific SVGs
[11]:
mcube_object <- readRDS(
file = file.path(
RESULT_PATH, seg_method,
paste0(
"mcube_object",
".rds"
)
)
)
sig_genes_list <- mcubeGetSigGenes(mcube_object@pvalues)
mcubeGetSigGenes: Set adjust_method as BH and alpha as 0.05.
[12]:
# According to nicheDE tutorial, the niche cell type is unknown when the resoluation is "CT" (cell type).
niche_genes_T <- get_niche_DE_genes(
NDE_obj, test.level = "CT",
index = "DCIS 2", niche = "Invasive Tumor",
pos = TRUE, alpha = 0.05
)$Genes
niche_genes_F <- get_niche_DE_genes(
NDE_obj, test.level = "CT",
index = "DCIS 2", niche = "Invasive Tumor",
pos = FALSE, alpha = 0.05
)$Genes
[1] "Returning Niche-DE Genes"
[1] "Returning Niche-DE Genes"
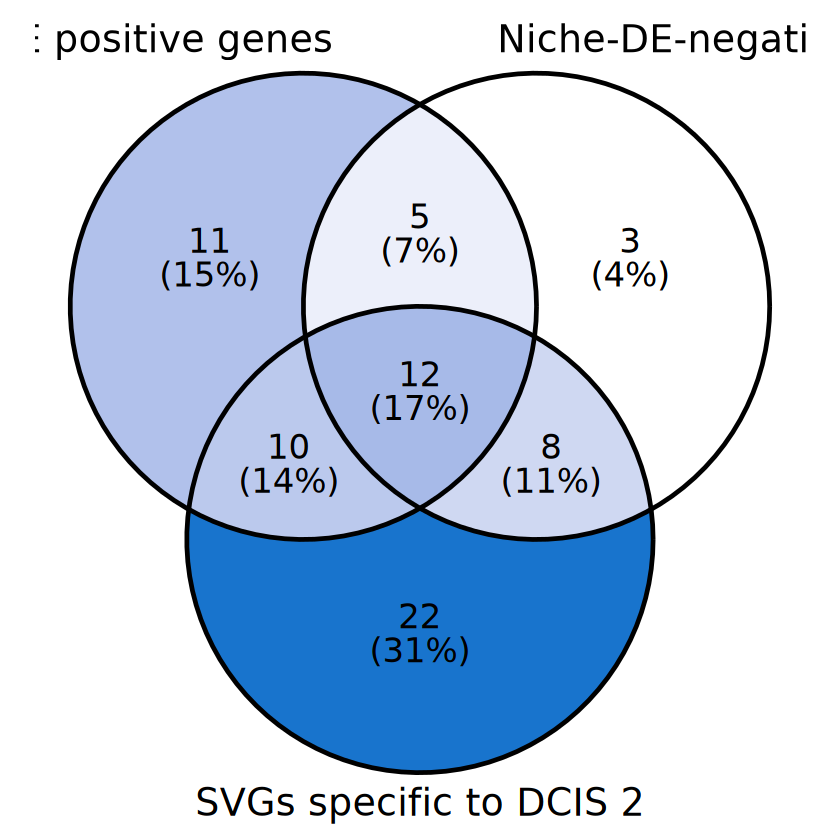
[13]:
library(ggVennDiagram)
celltype <- "DCIS 2"
svg_list <- list(niche_genes_T, niche_genes_F, rownames(sig_genes_list[[celltype]]))
name_list <- c(
"Niche-DE positive genes", "Niche-DE-negative genes",
paste0("SVGs specific to ", celltype)
)
p <- ggVennDiagram(svg_list, name_list, set_size = 8, label_size = 7, label_alpha = 0) +
scale_fill_gradient(low = "white", high = "dodgerblue3") +
# scale_fill_distiller(palette = "RdBu") +
theme(
text = element_text(size = 32),
title = element_text(size = 24),
legend.position = "none"
)
ggsave(
filename = file.path(RESULT_PATH, "niche_DE_genes_venn.pdf"),
plot = p, width = 10, height = 6
)
p
Attaching package: ‘ggVennDiagram’
The following object is masked from ‘package:spacexr’:
process_data
[ ]: